Oncomedicine 2016; 1:28-34. doi:10.7150/oncm.16930 This volume Cite
Review
A Focus on Current Molecular Pathways in Head and Neck
Ilias Karapantzos1, Paul Zarogoulidis2 ![]() , Michail Karanikas3, Vasilis Thomaidis4, Charalampos Charalampidis5, Chrysa Karapantzou1
, Michail Karanikas3, Vasilis Thomaidis4, Charalampos Charalampidis5, Chrysa Karapantzou1
1. Ear, Nose and Throat Department, “Saint Luke” Private Hospital, Panorama, Thessaloniki, Greece;
2. Pulmonary Department-Oncology Unit, “G. Papanikolaou” General Hospital, Aristotle University of Thessaloniki, Thessaloniki, Greece;
3. General Surgery Department, “Genisis” Private Clinic, Thessaloniki, Greece;
4. Department of Maxillofacial Surgery, University General Hospital of Alexandroupolis, Alexandroupolis, Greece;
5. Department of Anatomy, Democritus University of Thrace, Alexandroupolis, Greece.
Received 2016-7-22; Accepted 2016-8-19; Published 2016-11-18
Abstract
Early stages of head and neck cancer require concomitant administration of chemotherapy and radiotherapy. Currently platinum, taxane and fluoruracil analogs are being administered. On the other hand, for advanced stages extensive surgery is required. During the last years, several molecular characteristics have been identified in the tissue of head and neck squamous cell carcinoma, and novel treatment options are being pursued. In this review we will elucidate these molecular pathways that could be used as a possible treatment.
Keywords: head and neck cancer, chemotherapy, radiotherapy, molecular pathways.
Introduction
Treatment of head and neck squamous cell carcinoma (HNSCC) in early stages a requires non-invasive approach with concomitant chemotherapy and radiotherapy. There are still new data to come from ongoing studies whether taxane analog have better efficiency compared to platinum analogs [1]. Advanced stages require extensive surgery. Unfortunately, HNSCC of head and neck cancer has substantial heterogeneity, at the clinical and molecular level therefore it cannot be considered as a as a single disease entity. Currently two large studies investigate the mutational landscape of HNSCC [2, 3] as well as the pending release of data from the Cancer Genome Atlas Project (TCGA). This new information will shed light to novel therapies.
EGFR: New Data and Best Use of Inhibitors
Epidermal growth factor receptor (EGFR) was the first molecular pathway to be investigated. EGFR is a member of a family of receptor tyrosine kinases whose other members include ERBB2, ERBB3, and ERBB4. This pathway is responsible for the regulation of host of cellular activities including cell division, differentiation, and migration [4]. It has been demonstrated that EGFR overexpression is an early and very frequent molecular change in HNSCC, and the intensity of its expression is associated with reduced survival [5, 6]. EGFR is targeted either by small molecule adenosine-triphosphate competitive tyrosine kinase inhibitors (TKIs) specific to EGFR which bind to cytoplasmic region, such as gefitinib, erlotinib, or lapatinib, or by monoclonal antibodies (mAbs) directed at the extracellular domain of EGFR [7]. Cetuximab is the only U.S. FDA-approved targeted therapy available for HNSCC. Cetuximab is a chimeric IgG1-human monoclonal antibody against the extracellular domain of EGFR, which block the ligand binding to the receptor. Initial studies which evaluated cetuximab in human cancer cell lines in vitro and human tumor xenografts in vivo showed its potent antitumor activity [8]. Cetuximab involves several mechanisms, such as; induction of apoptosis, including inhibition of cell cycle progression, inhibition of metastasis, inhibition of angiogenesis, and its ability to enhance the response to chemotherapy and radiation [9]. The FDA has previously approved the addition of cetuximab to radiation for the treatment of locally advanced HNSCC. Cetuximab has been approved as a single agent for the treatment of patients with recurrent or metastatic HNSCC for patients whom platinum-based therapy has failed. Cetuximab has been administered with radiotherapy and was compared with radiotherapy alone in a multinational randomized study. During a 54 month follow up, the median survival time was 49 months in patients treated with cetuximab plus radiotherapy compared with 29.3 months in those treated with radiotherapy alone (p = 0.03) [10]. A 5-year evaluation of the overall survival results was conducted. The addition of cetuximab to radiotherapy significantly improved the survival of patients with HNSCC (p = 0.018) [11]. To date cisplatin chemoradiotherapy is considered the best choice for locally advanced, unresectable HNSCC, since no direct comparison between cetuximab radiotherapy and cisplatin chemoradiotherapy has been performed. Cetuximab currently is also being studied as a substitute to cisplatin in locally advanced HPV-associated oropharyngeal cancers (OSCC). Moreover; there two ongoing phase III studies which compare cetuximab with intensity modulated radiation therapy (IMRT) with cisplatin chemoradiotherapy in HPV-associated locally advanced oral squamous cell carcinoma [12]. In another phase II study The Eastern Cooperative Oncology Group (ECOG) (E1308) evaluated whether the increased response to platinum-based induction chemotherapy could be used to select patients who can then safely receive a lower dose of intensity-modulated radiation therapy (IMRT).
There are data in in vitro studies with HNSCC cell lines that show a synergistic effect of cetuximab with cisplatin [13]. Moreover; clinical data in the recurrent/metastatic setting confirmed the activity of this combination [14, 15]. However; there is the lack of survival from the addition of cetuximab to cisplatin chemoradiation because possibly cetuximab and cisplatin share the same mechanism of action. Therefore, it has proposed that other agents such as docetaxel might be better partners to cetuximab/radiotherapy.
Furthermore, administration of cetuximab has presented improved outcome of chemotherapy in the recurrent/metastatic setting. In another study it was reported that improved response rates were observed with the addition of cetuximab to cisplatin in ECOG 5397 [16]. In the study, conducted by Vermorken and colleagues, 442 eligible patients with previously untreated recurrent or metastatic HNSCC were randomly assigned it was observed that the addition of cetuximab to platinum-based chemotherapy with fluorouracil resulted in improvement in median OS from 7.4 to 10.1 months (p = 0.04). Moreover; median PFS was significantly prolonged from 3.3 to 5.6 months (p < 0.001), and the response rate increased from 20% to 36% (p < 0.001).
Furthermore, administration panitumumab which is a fully human monoclonal antibody against EGFR has been administered. Until now, there have been two phase II randomized studies, CONCERT 1 and 2, have been conducted in the unresectable locally advanced setting. Regarding CONCERT 1[17], it is a randomized phase II study of chemoradiotherapy with and without panitumumab. Regarding CONCERT 2 it is a randomized comparison of panitumumab radiotherapy to cisplatin chemoradiotherapy. However; both studies did not demonstrate superiority of the panitumumab arm, in specific it was observed that the addition of panitumumab to cisplatin chemoradiotherapy was associated with increased toxicity. Moreover; in the SPECTRUM study, which is a randomized phase III trial of chemotherapy with cisplatin/5-fluorouracil with or without panitumumab OS was not met. Until now EGFR TKIs have been tested in HNSCC but none have received FDA approval. In a recent phase II trial patients with recurrent or metastatic HNSCC received gefitinib, however; an overall response rate was only 11% [18]. Another a phase II study evaluated the association between adaptive dose-escalation to skin toxicity in the recurrent/metastatic setting and disease response [19]. Dose escalation was not associated with increased activity. In a phase III randomized study, the ECOG-E1302, docetaxel was compared to docetaxel plus gefitinib in patients with recurrent or metastatic HNSCC with PS 2 or who had been previously treated. The study was terminated early, because no statistically significant improvement in response rate, PFS, or OS compared with the docetaxel arm was observed. In another phase I study chemoradiotherapy was administered with lapatinib, a dual inhibitor which also disrupts the HER2 pathway, for locally advanced HNSCC reported an overall response rate of 81% [20]. Lapatinib has been observed to have little activity in EGFR inhibitor-naïve or -refractory subsets. Heterodimerization of EGFR with other HER family members are involved in resistance to EGFR inhibitors. Afatinib has a broad activity against multiple receptors in the HER family, making it theoretically more effective against tumor cells bearing several ErbB family members and heterodimerizations. In another, study a randomized phase II study compared weekly cetuximab (400 mg/m2 loading dose and 250 mg/m2 thereafter) with 50 mg of afatinib daily in 74 patients with recurrent or metastatic HNSCC in whom platinum-based therapy had failed [21]. Initial safety results analysis showed a side effect profile consistent with other EGFR inhibitors, with diarrhea and rash. It was observed that afatinib has activity in patients with recurrent/metastatic HNSCC in whom platinum-based therapy has failed and compares favorably to cetuximab. The major problem in EGFR-targeted therapy in HNSCC is patient selection, since the basic mechanism of resistance has not been identified. Regarding non-small cell lung cancer (NSCLC), patients with activating mutations in the EGFR tyrosine kinase domain demonstrate significantly higher response to EGFR TKIs (erlotinib, gefitinib and afatinib). However; mutations in EGFR tyrosine kinase domain are rare in HNSCC. There are several investigators reporting detection of the type III mutated variant (EGFRvIII) in a variable proportion of HNSCC ranging from 0% to 40%, [22] however; there are still data to be clarified.
EGFRvIII mutated variant is characterized by an in-frame deletion from exons 2 through 7 in the extracellular domain irreversible EGFR inhibitors such as afatinib in preclinical studies have been found to be effective against EGFRvIII.
It has been observed that overexpression of ERBB2-induced gefitinib resistance, and also the combination of pertuzumab (which targets HER2) with gefitinib in HNSCC cell lines resistant to gefitinib resulted in increased inhibition of cell growth. [23] It has been observed that mutations in KRAS are extremely rare in HNSCC, however; HRAS mutations have been reported [2].
Another molecular pathway the MET overexpression was found in 84% of HNSCC cases; The MET mutations and gene amplification have been reported in 13.5% and 13% of the cases, respectively. It was also found that a combination of a MET inhibitor with erlotinib resulted in greater-than-additive inhibition of cell growth by ErbB3/AKT signaling [24].
Future research will elucidate the factors that correlate with response of EGFR-targeted therapies.
Phosphatidylinositide 3-Kinase Pathway Biology and Inhibitors
The PI3K-AKT-mTOR Pathway and Physiology
Phosphoinositide 3-kinases (PI3Ks) are a family of related enzymes that play a pivotal role to the cellular regulatory mechanisms. It has been observed that their function has been linked to the regulation of numerous biologic processes, including cell growth, migration, survival, proliferation, differentiation, and differentiation [25, 26]. According to structural and substrate specificity, PI3Ks are divided into 3 classes. Class 1 is currently the only clinically relevant subgroup and can be further divided into A and B subtypes: Class 1 A, Class 1 B enzymes, Class 2 and Class 3. PI3-kinases are linked with several cellular functions such as; cell growth, differentiation, motility, proliferation, survival, and intracellular trafficking. Several of these functions are related to the ability of class 1 PI3-kinases to activate protein kinase B (PKB, also commonly known as AKT). AKT, is a serine-threonine kinase, which has three different isoforms (AKT1, AKT2, AKT3). AKT activation is known to be initiated by translocation to the plasma membrane. AKT phosphorylation is carried out by 3-phosphoinositide-dependent protein kinase 1 (PDK1) and PDK2. It is currently thought that mTOR-rictor (rapamycin insensitive companion of mTOR) complex 2 (mTORC2) is the primary source of PDK2 activity under most circumstances.
It is known that there are several AKT-independent pathways, however; their role in cancer is not well defined. Upon AKT activation which is a phosphorylating source for many proteins, including glycogen synthase phosphate 3 and FOXOs, several diverse cellular functions are regulated. It is known that in most cancers, AKT activation by PI3K is either by tyrosine kinases (RTKs) or by somatic mutations [25, 26]. It has been found that PI3K-AKT signaling plays diverse role in normal and cancer physiology through multiple downstream pathways.
PTEN loss
It is known that PTEN functionally antagonizes PI3K activity through its intrinsic lipid phosphatase activity that reduces the cellular pool of PtdIns [3,4,5] P3 by converting PtdIns [3,4,5] P3 back to phosphatidylinositol-4,5-bisphosphate (PtdIns [4,5 ]P2). It has been observed that loss of PTEN results in unrestrained signaling by the PI3K pathway, which leads to cancer. It has been observed that PTEN loss is common in head and neck cancer [25, 27]. It is known that tumors with PTEN loss appear to signal primarily using the p110beta (PIK3CB), this information is vital for the selection of appropriate inhibitors [28]. The loss of PTEN tumor suppressor is common, although mutations may not be the primary mechanism of PTEN loss in HNSCC [29].
Genetic Alterations in the PI3K Pathway in Head and Neck Cancer
PIK3CA mutations
It has been observed that that there are genetic aberrations of the PI3K pathway in head and neck cancer. These mutations of the PIK3CA gene have been reported in 6% to 20% of head and neck tumors [2, 29]. Moreover; it has been observed that there are well-established oncogenic (canonical) PIK3CA mutations in HPV-positive head and neck cancers [30].
There are also other genetic mutations such as; PIK3R1 mutations, AKT mutations and amplification, TSC1/2 or PDK1 aberrations, INPP4/PHLPP tumor suppressor loss, or loss of NF2 [25, 26]. However; it remains unclear whether these genetic mutations occur in HNSCC. Regarding epidermal growth factor which is amplified in 10% to 15% of HNSCC, [29] signaling may not solely rely on PI3K [25, 26].
Translational Implications
It has been observed in vitro that PI3K mutations may induce increased resistance to EGFR inhibition [31]. PIK3CA may be biomarker for EGFR resistance.
Pi3k Inhibitors in Clinical Testing and Trials in Hnscc
There are three classes of PI3K inhibitors that can be differentiated: Combined inhibitors of PI3K/mTOR, Pan-Class I PI3K inhibitors, Alpha-specific (p110alpha specific) inhibitors.
It remains unclear which type of PI3K inhibitors could be the most promising, however; the vast genetic aberration in the PI3K pathway and early clinical data indicate that PI3K could be a promising target for HNSCC [27].
New Signaling Pathways and Targets in the Hnscc Horizon
NOTCH
There is also the NOTCH family which consists of four receptors (NOTCH1-4), which interact with the Delta-like (Dll1, Dll3 and Dll4) and Jagged (Jag1 and Jag2) families of ligands. These are normally bound to the cell membrane. It is known that ligand binding is followed by two cleavages of the NOTCH receptor by ADAM metalloprotease and gamma-secretase complex, leading to the release of the NOTCH intracellular domain (NICD). It has been observed that NICD is associated with the CSL/MAM complex which is able to bind DNA and promote transcription [32]. NOTCH signaling pathway is known to be a pro-tumorigenic pathway. When activated them mutations and translocations are observed in the genes for NOTCH receptors or their regulators. NOTCH1 heterodimerization (HD) domain mutations can result in ligand independent proteolytic activation which then drive proliferation in T-ALL [33]. Furthermore, these activating mutations result in HES 1 upregulation, which in turn induce suppression of transcription of PTEN. It has been previously observed that PTEN loss is a gateway to resistance to NOTCH1 inhibition in T-ALL [34].
Moreover; several groups with the use of next-generation exome sequencing have observed that NOTCH mutations occur second in frequency only to TP53 mutations in head and neck, lung squamous cell carcinomas (SCC) and skin [35].
Currently there is an interest in NOTCH signaling in HNSCC because identifying the pathways that become dysregulated after NOTCH inactivation is essential in order to identify effectiveness of a treatment approach.
ALK1
It is known that activin receptor-like kinase 1 (ALK1) is a type I receptor belonging to the transforming growth factor-beta (TGFβ) superfamily. This protein is selectively expressed on activated endothelial cells in response to injury or disease [36]. ALK1 has been hypothesized to play a key role in the development of functional vasculature.
ACE-041 protein binds to the ligands bone morphogenetic protein (BMP) 9 and BMP10 and inhibits their interaction with ALK1, and finally blocks ALK1-mediated signaling. ACE-041 binding disrupts the process of vascular development [37]. ACE-041 has been studied in a dose-escalation phase I study in order to determine its safety profile, optimal dosing strategy, tolerability, and antitumor activity in patients with advanced solid tumors [37]. The main toxicity observed was edema and fluid overload that were dose-dependent and responded to diuretic therapy. Based on the favorable outcome of this study currently there is an ongoing phase II clinical trial of ACE-041 in patients with refractory relapsed/metastatic (NCT01458392).
Hedgehog Pathway
It is known that the hedgehog pathway (HhP) is a validated anticancer target, and vismodegib (a small molecule that inhibits the HhP) is used by patients with advanced basal cell carcinoma of the skin. Ligand activation by sonic hedgehog (SHH) leads to a cascade of signaling, which in turn modulates the expression of numerous cancer target genes. HhP signaling is also crucial for drug resistance [38, 39]. To date it has been observed that EGFR and HhP are implicated as key drivers of proliferation and survival of cancer cells.
Moreover, it has been observed that chronic gefitinib treatment generates a mesenchymal drug-resistant population in HNSCC cells independent of EGFR activation [40]. However; this dichotomy of EGFR-dependent population and -resistant states and the role of HhP signaling have not been yet clarified in HNSCC.
It has been observed that GLI1 is a key driver of tumor growth and metastasis in multiple cancers [41, 42]. Regarding head and neck squamous cell carcinoma nuclear GLI1 expression levels were determined in tumors from patients enrolled on RTOG 9003, a radiation fractionation trial [43]. In this study the results were also correlated with previously determined EGFR expression, and assessed in relation to time to metastasis (TTM). It was observed that GLI1 was associated with poorer outcomes, tumor/node/metastasis staging system stages, adjusted for age, and performance status. In this study, data suggested that GLI1 could serve as a marker in HNSCC, however, since several pathways other than HhP converge modulate GLI1, and thus the regulatory mechanisms, the significance of these findings are yet to be clarified.
Currently based on preclinical data using a vast variety of in vitro and patient-derived in vivo models of HNSCC and the HhP inhibitor IPI-926, [44] cetuximab and IPI-926 (NCT01255800) are being administered with a dose escalation concept. In this study the authors investigate the transcriptome analysis in order to determine the molecular mechanisms underlying EGFR acquired resistance and how HhP modulation may modulate such EGFR dependence. Table 1, Figure 1.
Current selected targeted agents from www.clinicaltrial.gov.
| Drug Name (Tradename) Target(s) Phase of study in Head and Neck cancer | |
|---|---|
| Cetuximab (Erbitux) EGFR | III |
| Gefitinib (Iressa) EGFR | I/II/III |
| Sunitinib (Sutent) VEGF | I/II |
| Bevacizumab (Avastin) VEGF | I/II/III |
| Romidepsin Histone deacetylase | I/II |
| Vorinostat (Zolinza) Histone deacetylase | I/II |
| Dasatinib (Sprycel) Tyrosine kinases | II |
| Imatinib (Gleevec) Tyrosine kinases | II |
| Pazopanib VEGF, Tyrosine kinases | II |
| Vandetanib (Zactima) VEGF, EGFR | I/II |
| XL880 VEGF, Tyrosine kinases | II |
| Perifosine (KRX-0401) Akt | II |
| Bortezomib (Velcade) NF-kB, Tyrosine kinases | I/II |
| Lonafarnib (Serasar) Farnesyl transferase | I/II |
| Tanespimycin (KOS-953) Hsp90 | I/II |
| AZD0530 Src/Abl kinase | II |
| Erlotinib (Tarceva) EGFR | I/II/III |
| Panitumumab (Vectibix) EGFR | I/II/III |
| BIBW 2992 (Tovok) EGFR, HER-2/neu | II |
| Zalutumumab (HuMax-EGFr) EGFR | I/II/III |
| Trastuzumab (Herceptin) HER-2/neu | II |
| Lapatinib (Tykerb) EGFR, HER-2/neu | I/II/III |
| Cediranib (Recentin) VEGF | I/II |
| Sorafenib (Nexavar) Raf, VEGF | I/II |
| Semaxanib VEGF | I/II |
| Pazopanib VEGF | II |
Most important molecular pathways for Head and Neck cancer.
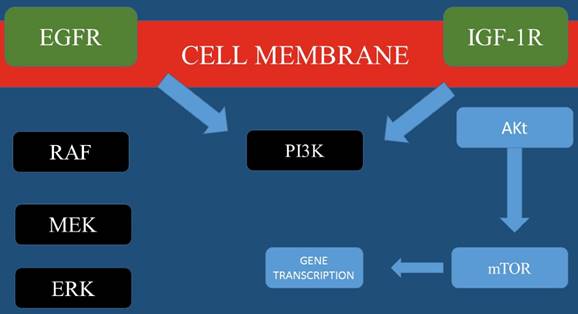
In conclusion, currently there are many novel pathways and treatment approaches which are being explored for head and neck cancer. These pathways include EGFR activity and methods to overcome resistance and targeting PI3K signaling. Moreover; identifying new candidate treatment targets such as ALK1, NOTCH, and hedgehog signaling.
Competing Interests
None to declare.
References
1. Wang CJ, Knecht R. Current concepts of organ preservation in head and neck cancer. European archives of oto-rhino-laryngology: official journal of the European Federation of Oto-Rhino-Laryngological Societies. 2011;268:481-7 doi:10.1007/s00405-010-1407-8
2. Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A. et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157-60 doi:10.1126/science.1208130
3. Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ. et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011;333:1154-7 doi:10.1126/science.1206923
4. Schlessinger J. Common and distinct elements in cellular signaling via EGF and FGF receptors. Science. 2004;306:1506-7 doi:10.1126/science.1105396
5. Grandis JR, Tweardy DJ. Elevated levels of transforming growth factor alpha and epidermal growth factor receptor messenger RNA are early markers of carcinogenesis in head and neck cancer. Cancer research. 1993;53:3579-84
6. Grandis JR, Tweardy DJ. TGF-alpha and EGFR in head and neck cancer. Journal of cellular biochemistry Supplement. 1993;17F:188-91
7. Egloff AM, Grandis J. Epidermal growth factor receptor-targeted molecular therapeutics for head and neck squamous cell carcinoma. Expert opinion on therapeutic targets. 2006;10:639-47 doi:10.1517/14728222.10.5.639
8. Harari PM, Huang SM. Head and neck cancer as a clinical model for molecular targeting of therapy: combining EGFR blockade with radiation. International journal of radiation oncology, biology, physics. 2001;49:427-33
9. Harari PM, Huang SM. Radiation response modification following molecular inhibition of epidermal growth factor receptor signaling. Seminars in radiation oncology. 2001;11:281-9
10. Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM, Cohen RB. et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. The New England journal of medicine. 2006;354:567-78 doi:10.1056/NEJMoa053422
11. Bonner JA, Harari PM, Giralt J, Cohen RB, Jones CU, Sur RK. et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. The Lancet Oncology. 2010;11:21-8 doi:10.1016/S1470-2045(09)70311-0
12. Ang KK, Harris J, Wheeler R, Weber R, Rosenthal DI, Nguyen-Tan PF. et al. Human papillomavirus and survival of patients with oropharyngeal cancer. The New England journal of medicine. 2010;363:24-35 doi:10.1056/NEJMoa0912217
13. Takaoka S, Iwase M, Uchida M, Yoshiba S, Kondo G, Watanabe H. et al. Effect of combining epidermal growth factor receptor inhibitors and cisplatin on proliferation and apoptosis of oral squamous cell carcinoma cells. International journal of oncology. 2007;30:1469-76
14. Vermorken JB, Herbst RS, Leon X, Amellal N, Baselga J. Overview of the efficacy of cetuximab in recurrent and/or metastatic squamous cell carcinoma of the head and neck in patients who previously failed platinum-based therapies. Cancer. 2008;112:2710-9 doi:10.1002/cncr.23442
15. Ang KK, Zhang Q, Rosenthal DI, Nguyen-Tan PF, Sherman EJ, Weber RS. et al. Randomized phase III trial of concurrent accelerated radiation plus cisplatin with or without cetuximab for stage III to IV head and neck carcinoma: RTOG 0522. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2014;32:2940-50 doi:10.1200/JCO.2013.53.5633
16. Burtness B, Goldwasser MA, Flood W, Mattar B, Forastiere AA, Eastern Cooperative Oncology G. Phase III randomized trial of cisplatin plus placebo compared with cisplatin plus cetuximab in metastatic/recurrent head and neck cancer: an Eastern Cooperative Oncology Group study. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2005;23:8646-54 doi:10.1200/JCO.2005.02.4646
17. Giralt J FA, Mesia R. et al. A phase II, randomized trial (CONCERT-1) of chemoradiotherapy (CRT) with or without panitumumab (pmab) in patients (pts) with unresected, locally advanced squamous cell carcinoma of the head and neck (LASCCHN). J Clin Oncol. 2012;30(suppl):abstr5502
18. Chua DT, Wei WI, Wong MP, Sham JS, Nicholls J, Au GK. Phase II study of gefitinib for the treatment of recurrent and metastatic nasopharyngeal carcinoma. Head & neck. 2008;30:863-7 doi:10.1002/hed.20792
19. Perez CA, Song H, Raez LE, Agulnik M, Grushko TA, Dekker A. et al. Phase II study of gefitinib adaptive dose escalation to skin toxicity in recurrent or metastatic squamous cell carcinoma of the head and neck. Oral oncology. 2012;48:887-92 doi:10.1016/j.oraloncology.2012.03.020
20. Harrington KJ, El-Hariry IA, Holford CS, Lusinchi A, Nutting CM, Rosine D. et al. Phase I study of lapatinib in combination with chemoradiation in patients with locally advanced squamous cell carcinoma of the head and neck. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2009;27:1100-7 doi:10.1200/JCO.2008.17.5349
21. Seiwert TY HD, Cohen EE. et al. A randomized phase II trial of cetuximab-based induction chemotherapy followed by concurrent cetuximab, 5-FU, hydroxyurea, and hyperfractionated radiation (CetuxFHX), or cetuximab, cisplatin, and accelerated radiation with concomitant boost (CetuxPX) in patients with locoregionally advanced head and neck cancer (HNC). J Clin Oncol. 2011;29(suppl):abstr5519
22. Sok JC, Coppelli FM, Thomas SM, Lango MN, Xi S, Hunt JL. et al. Mutant epidermal growth factor receptor (EGFRvIII) contributes to head and neck cancer growth and resistance to EGFR targeting. Clinical cancer research: an official journal of the American Association for Cancer Research. 2006;12:5064-73 doi:10.1158/1078-0432.CCR-06-0913
23. Erjala K, Sundvall M, Junttila TT, Zhang N, Savisalo M, Mali P. et al. Signaling via ErbB2 and ErbB3 associates with resistance and epidermal growth factor receptor (EGFR) amplification with sensitivity to EGFR inhibitor gefitinib in head and neck squamous cell carcinoma cells. Clinical cancer research: an official journal of the American Association for Cancer Research. 2006;12:4103-11 doi:10.1158/1078-0432.CCR-05-2404
24. Seiwert TY, Jagadeeswaran R, Faoro L, Janamanchi V, Nallasura V, El Dinali M. et al. The MET receptor tyrosine kinase is a potential novel therapeutic target for head and neck squamous cell carcinoma. Cancer research. 2009;69:3021-31 doi:10.1158/0008-5472.CAN-08-2881
25. Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nature reviews Cancer. 2009;9:550-62 doi:10.1038/nrc2664
26. Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nature reviews Drug discovery. 2009;8:627-44 doi:10.1038/nrd2926
27. Holsinger FC, Piha-Paul SA, Janku F, Hong DS, Atkins JT, Tsimberidou AM. et al. Biomarker-directed therapy of squamous carcinomas of the head and neck: targeting PI3K/PTEN/mTOR pathway. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2013;31:e137-40 doi:10.1200/JCO.2012.43.2716
28. Wee S, Wiederschain D, Maira SM, Loo A, Miller C, deBeaumont R. et al. PTEN-deficient cancers depend on PIK3CB. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:13057-62 doi:10.1073/pnas.0802655105
29. Morris LG, Taylor BS, Bivona TG, Gong Y, Eng S, Brennan CW. et al. Genomic dissection of the epidermal growth factor receptor (EGFR)/PI3K pathway reveals frequent deletion of the EGFR phosphatase PTPRS in head and neck cancers. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:19024-9 doi:10.1073/pnas.1111963108
30. Seiwert TY KM, Zuo Z. et al. Genomic profiling of a clinically annotated cohort of locoregionally advanced head and neck cancers (HNC) treated with definitive chemoradiotherapy. J Clin Oncol. 2012;30(suppl):abstr5517
31. Young NR, Liu J, Pierce C, Wei TF, Grushko T, Olopade OI. et al. Molecular phenotype predicts sensitivity of squamous cell carcinoma of the head and neck to epidermal growth factor receptor inhibition. Molecular oncology. 2013;7:359-68 doi:10.1016/j.molonc.2012.11.001
32. Bray SJ. Notch signalling: a simple pathway becomes complex. Nature reviews Molecular cell biology. 2006;7:678-89 doi:10.1038/nrm2009
33. Malecki MJ, Sanchez-Irizarry C, Mitchell JL, Histen G, Xu ML, Aster JC. et al. Leukemia-associated mutations within the NOTCH1 heterodimerization domain fall into at least two distinct mechanistic classes. Molecular and cellular biology. 2006;26:4642-51 doi:10.1128/MCB.01655-05
34. Palomero T, Sulis ML, Cortina M, Real PJ, Barnes K, Ciofani M. et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nature medicine. 2007;13:1203-10 doi:10.1038/nm1636
35. Wang NJ, Sanborn Z, Arnett KL, Bayston LJ, Liao W, Proby CM. et al. Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:17761-6 doi:10.1073/pnas.1114669108
36. Seki T, Yun J, Oh SP. Arterial endothelium-specific activin receptor-like kinase 1 expression suggests its role in arterialization and vascular remodeling. Circulation research. 2003;93:682-9 doi:10.1161/01.RES.0000095246.40391.3B
37. Cunha SI, Pietras K. ALK1 as an emerging target for antiangiogenic therapy of cancer. Blood. 2011;117:6999-7006 doi:10.1182/blood-2011-01-330142
38. Liu S, Dontu G, Mantle ID, Patel S, Ahn NS, Jackson KW. et al. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer research. 2006;66:6063-71 doi:10.1158/0008-5472.CAN-06-0054
39. Feldmann G, Dhara S, Fendrich V, Bedja D, Beaty R, Mullendore M. et al. Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: a new paradigm for combination therapy in solid cancers. Cancer research. 2007;67:2187-96 doi:10.1158/0008-5472.CAN-06-3281
40. Maseki S, Ijichi K, Tanaka H, Fujii M, Hasegawa Y, Ogawa T. et al. Acquisition of EMT phenotype in the gefitinib-resistant cells of a head and neck squamous cell carcinoma cell line through Akt/GSK-3beta/snail signalling pathway. British journal of cancer. 2012;106:1196-204 doi:10.1038/bjc.2012.24
41. Thayer SP, di Magliano MP, Heiser PW, Nielsen CM, Roberts DJ, Lauwers GY. et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003;425:851-6 doi:10.1038/nature02009
42. Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i Altaba A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Current biology: CB. 2007;17:165-72 doi:10.1016/j.cub.2006.11.033
43. Chung CH, Dignam JJ, Hammond ME, Klimowicz AC, Petrillo SK, Magliocco A. et al. Glioma-associated oncogene family zinc finger 1 expression and metastasis in patients with head and neck squamous cell carcinoma treated with radiation therapy (RTOG 9003). Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2011;29:1326-34 doi:10.1200/JCO.2010.32.3295
44. Mimeault M, Johansson SL, Vankatraman G, Moore E, Henichart JP, Depreux P. et al. Combined targeting of epidermal growth factor receptor and hedgehog signaling by gefitinib and cyclopamine cooperatively improves the cytotoxic effects of docetaxel on metastatic prostate cancer cells. Molecular cancer therapeutics. 2007;6:967-78 doi:10.1158/1535-7163.MCT-06-0648
Author contact
![]() Corresponding author: Paul Zarogoulidis, M.D, PhD. Pulmonary Department-Oncology Unit, “G. Papanikolaou” General Hospital, Aristotle University of Thessaloniki, Thessaloniki, Greece. Fax: 00302310992424 Mobile: 00306977271974 E-mail: pzarogcom.
Corresponding author: Paul Zarogoulidis, M.D, PhD. Pulmonary Department-Oncology Unit, “G. Papanikolaou” General Hospital, Aristotle University of Thessaloniki, Thessaloniki, Greece. Fax: 00302310992424 Mobile: 00306977271974 E-mail: pzarogcom.