Oncomedicine 2017; 2:42-51. doi:10.7150/oncm.16758 This volume Cite
Review
Base Excision Repair Manipulation in Breast Carcinoma: A Prospective Avenue to Potentiate Genome Insulting Approach
Aayushi Jain, Sunny Yadav, Mohsein Beig, Shruti Purohit, Nilesh Kumar Sharma ![]()
Cancer and Translational Research Lab, Dr. D.Y Patil Biotechnology & Bioinformatics Institute, Dr. D. Y. Patil Vidyapeeth, Pune, Maharashtra, India, 411033.
Received 2016-7-7; Accepted 2016-12-6; Published 2017-2-22
Abstract
Treating breast carcinoma gets tedious due to its invasive molecular subtypes and their various molecular genotypes. Depending upon genotype and phenotype of breast tumor types, they manipulate their survival arms in the form of DNA repair protein player including base excision repair (BER) pathway. Currently, avenues to treat breast cancer ate genotoxic drugs inflicting inter and intra-strand cross links, base modification and changes in the genome combined with inhibitors of BER pathway. This review summarizes the updated information on the relevance of BER response in breast carcinoma phenotypes and their potential therapeutic interference in the last decade.
Keywords: Base excision repair, Breast carcinoma, Chemotherapy, DNA damage response, Genotoxic Drug, Genomic instability, Radiation therapy
Introduction
Breast carcinoma, a heterogeneous cancer is caused due to various entities comprising genotype, functionality, reproductive, environmental and lifestyle. The clinical behavioral changes of the breast carcinoma cell types arise from the change in their genetic aberrations, epigenetic modifications and transcriptional regulation [1-6]. In growing evidences, breast carcinoma and other types of cancer are linked with the swinging balance between DNA damage build up and DNA repair failure. The DNA repair umbrella in breast cancer involves several molecular players and their set basic signal transduction pathway [4-6]. These set of DNA damage response players are often found to act as tumor suppressor and hyperactive repair systems to confer survival advantage to breast cancer against genomic insults [7].
Currently, the problem in genotoxic and radiation therapy responsiveness and resistance in breast carcinoma is highly witnessed. One of the plausible mechanisms behind above caveats is the role of abnormal and compensatory DNA repair among genotypically different breast carcinoma cells [8-12]. Chemotherapy and radiotherapy are widely opted mode of treatment for breast carcinoma but they do have setbacks [10, 11, 13, 14]. Genetic materials of the normal and breast carcinoma cells encode for purpose of multiplying and proper incorporation of functional proteins in development, proliferation and external insults survival. Besides outside genomic insults, replication and inherent process lead to many unintentional and unavoidable changes including incorrect nucleotide base incorporation or deletion, breakage of DNA strand and chemical modifications [10-12, 14-16]. There are growing thoughts that both normal and carcinoma cells instigate strategies to thwart such unfavorable genomic instability. These changes in the genetic material viciously play a role in wake up and surveillance in normal and breast carcinoma cells. Both type of cells work with programmed rules to check such modifications and damages by repair or removal process [10, 11, 17]. DNA repair machinery is an in-built weapon of the cell to maintain the genome integrity by repairing the damage, inducing apoptosis and leading to arrest in cell cycle [10, 11, 14, 17-20]. DNA damages are found due to many extracellular and intracellular chemical factors like free reactive oxygen species, base hydrolyzing agents or by exposure of cells to harmful rays. Rigorous accumulation of such mutations caused by DNA damage plays a role in cancer progression [10, 11, 14, 21, 22]. Several inhibitors/drugs/interference approaches are considered to target specific repair proteins of DNA repair pathways such as BER, nucleotide excision repair (NER), homologous recombination (HR) and non-homologous end joining (NHEJ) pathways in breast carcinoma [17,18,23,24]. In this review, the authors attempt to encapsulate status on promising potential of combinatorial therapy of chemo/radiotherapy and specific BER protein inhibitors in the last five years related to breast carcinoma.
DNA Repair Response and Breast Carcinoma Phenotypes
Breast carcinoma is classified as complex and heterogeneous disease [25]. Despite limitless efforts to combat breast carcinoma, there are rise of incidences in the past few years. The rising statistics of breast carcinoma among women are due to various factors like mutations, genetic factors, hormone imbalance, pregnancy in later age and late menopause [1-4, 6, 25-27]. The breast cancer phenotypes are mainly classified clinically. However, to emphasize the classification based upon the DNA damage repair genomic and proteomics look promising for future individualized therapy [26]. In recent, several studies are reported about breast cancers and neoplasms linked to DNA damage repair defects, cell-cycle checkpoints alterations and strategies to thwart genomic attack. It is widely viewed that modulation of DNA damage response pathways is commonly accepted strategies by most of breast cancer phenotypes [1, 6, 28].
The breast cancer is commonly classified according to hormone receptor (in particular estrogen and progesterone receptors) and epidermal growth factor receptor ErbB2/HER2 presence [1, 6, 26, 27]. According to this classification, breast carcinoma is named as hormone receptor positive; hormone receptor negative with HER2 over-expression; triple negative breast cancer which does not express any of the three receptors. Triple-negative breast cancers often contain inactivation of the DNA repair gene BRCA1 [29]. According to survey data, 30% of breast cancers are supposed to have some degree of BRCA1 inactivation, where triple-negative breast cancers are often detected with BRCA1 inactivation. It is accepted that hormone receptor positive cancers are preferably treated with hormone modulators drugs, radiation therapy and surgery. But, discernible bottleneck is the less response towards genomic insults due to the acquired survival strategy by breast carcinoma [1, 3, 25]. Specifically, triple-negative breast cancers (TNBC) referred as hormone receptor negative (hormone insensitive) and HER-2 negative display inactivation of BRCA1 and DNA repair defects [4, 25, 26, 27]. Besides tumor grade, molecular subtypes are preferentially used in the research settings, where Luminal A is classified as ER-positive and/or PR-positive, HER2-negative with Low Ki67. Another breast carcinoma molecular subtype is referred as Luminal B defined by ER-positive and/or PR-positive, HER2-positive (or HER2-negative with high Ki67) with prevalence around 10-20%. Another molecular subtype as Triple negative/basal-like is noted as ER-negative/PR-negative/HER2-negative with prevalence of around 15-20% [1, 3, 4, 6, 25-27]. Besides another molecular subtype as HER2posses ER-negative/PR-negative/HER2-positive characteristics with prevalence of 5-15%.
Several subtypes of breast cancer are recognized for their abnormalities in DNA damage repair and BRCA1 inactivation through mutation or epigenetic modification. Breast cancer phenotypes are broadly classified as inherited vs sporadic in nature. The genetic contributions to breast cancer are elucidated as presence of several autosomal dominant cancer syndromes mostly with highly penetrant germline mutations in BRCA1 and BRCA2 [4, 6, 26, 27]. Breast cancers around 5-10% are classified as hereditary breast carcinoma and known gene players such as BRCA1, BRCA2, CHEK2, BRIP1, RAD51 and ATM linked with breast cancers with moderate penetrance [6, 30]. Among the DNA repair response players, BRCA1 and BRCA2 are key players in homologous recombination type of error-free repair system. The BRCA1 and BRCA2 are known for their role in transcription, double strand break repair, recombination, tumor suppressor and maintenance of genomic stability [31]. In past decade, findings support that breast cancer due to germ line mutation in BRCA1 and BRCA2 genes display distinct morphology, immunophenotype and molecular characteristics from age-matched sporadic breast cancer cases [1, 3, 4, 6, 25, 26, 27].
BRCA1-associated cancers are often regarded as ER negative and triple negative (ER-, PgR-, HER2-). At the same time, a high proportion shows a 'basal' phenotype. The BRCA1-associated carcinomas are associated with the cell cycle proteins such as E2F6, cyclins A and SKP2 [31, 32]. The BRCA2 associated tumors are documented to show higher expression of the cell cycle proteins such as cyclin D1, cyclin D3, p27, p16, p21, CDK4, CDK2 and CDK1 [6,26,27]. The BRCA1 gene is associated with DNA repair, cell-cycle regulation, transcriptional regulation and epigenetic chromatin remodeling. On the other side, BRCA2 gene is devoted to DNA recombination and repair processes [1, 3, 6, 25, 26, 27]. It is documented that BRCA1 or BRCA2 loss result into lack of DNA double-strand breaks repair by the error-free mechanism of homologous recombination (HR). The deficiency in HR repair ensue compensatory and alternative error-prone, and potentially mutagenic, repair of DNA lesions by mechanisms such as non-homologous end joining and single-strand annealing [6, 26, 27].
The most common demarcation of breast cancer is based upon inherited susceptibility to breast cancer vs sporadic occurrences. It is widely accepted that genetic alterations increase susceptibility due to inherited heterozygous gene defect in BRCA1, TP53 and PTEN [1, 3, 25]. The supporting evidences substantiate that gene factors such as tumor suppressor genes associated with genome fidelity as BRCA1 (DNA damage repair), TP53 (cell cycle checkpoint) and PTEN (blockage of cell-cycle progression in G1 and participation in DNA repair). According to literature, 5%-10% of breast cancer cases are originated due to germ-line mutations, but similar genetic abnormalities observed in individuals without genetic pre-dispositions [1, 3, 4, 6, 25, 26, 27]. There is clear view that presence of an acquired mutation or epigenetic inactivation occurs in case of sporadic breast cancer comprises of key genes involved in DNA damage repair process.
Hereditary breast cancers are considered as a part of several cancer syndromes encompassing the Li-Fraumeni syndrome (TP53) [33] and Cowden's syndrome (PTEN) caused by high-penetrant genes. The hereditary breast cancer represents small contribution to familial breast cancer. The reason behind the manipulation and subversion of DNA repair response on breast carcinoma is explored in the perspectives of hormonal signaling. There are sound evidences that certain steroid hormones alter the double strand break repair mediated by BRCA1 in case of hereditary type of breast cancer [34]. In BRCA1 and BRCA2 mutation associated cancer, overexpression of HER2 is seldom. Conversely, BRCA1 tumors frequently show mutations in TP53. Such observations are not featured in BRCA2 cancers. Based on the previous genetic and pathological study, BRCA1-associated breast cancers are more likely to be estrogen (ER) and progesterone receptor (PgR) negative (~90%) matched with sporadic breast cancers (~30%). In contrast, the occurrence of ER and PgR for BRCA2- and BRCAX-associated cancers is not considerably different to sporadic cancers [1, 3, 6, 34].
By subverting HR based repair system, base excision and single-strand break repair system get started and help breast carcinoma to survive and elude external genotoxic threats. Therefore, there are perceptions and attempts made to impede these pathways in breast carcinoma types harboring BRCA mutation to produce loss of the repair mechanisms leading to cell death [1, 3, 25]. TNBC breast cancers tend to be sensitive to traditional chemotherapeutic agents and response may be due to their aberrant DNA damage responses generating massive spontaneous DNA damage and causing catastrophic chromosomal abnormalities in cancer cells with defective repair genes such as BRCA1 [4, 6, 25, 26, 27]. The normal and cancer cells employ defined pool DNA repair pathways specific for plethora of DNA lesions using dedicated and specialized proteins/enzymes. However, questions are interrogated to know the DNA repair response difference between hereditary and sporadic breast cancer. It is conceived that differential expression in hereditary breast cancer for DNA repair proteins as poly ADP-ribose polymerase (PARP) 1 (non-cleaved), BARD1 and RAD51 compared to sporadic breast cancer. DNA repair markers show differential expression in BRCA-mutated tumors with a substantial degree of disruption of DNA repair pathways in sporadic breast cancer [27].
BER and Their Molecular Actors
The DNA damage repair machinery of the both normal as well as breast carcinoma is responsible for maintenance of the genome integrity and stability along with needed manipulations during the state genomic insults imposed from outside [12, 14, 19]. The well-crafted DNA damage repair machinery inside the cell is tightly regulated and according the division of work inside the cellular periphery, a specific family of repair team agent is recruited when particular lesion is sensed. Onset of carcinoma changes the DNA damage response which can be the cause of enhanced genotoxic stress which can be considered as targeted therapeutic agents [34].
Normal and carcinoma cells utilize complex and well-regulated DNA damage response (DDR) pathways to protect genome from DNA damage induced by DNA replication, cellular metabolism and exogenous genomic insults. The repair pathways comprise several well-connected pathways to repair single-strand breaks (SSBs) through BER, DNA double strand break (DSBs) by means of either homologous recombination (HR) or non-homologous end-joining (NHEJ), bulky DNA adducts via nucleotide excision repair (NER) and mismatches/insertions-deletions via mismatch repair (MMR) [8, 35, 36, 37, 38]. Alterations in the DDR machinery increase cancer susceptibility by creating potentially transforming mutations, cell death. Understanding about DDR contribution to carcinogenesis and pinpointing for therapeutic benefit is highly warranted.
The DNA continuously gets damaged due to endogenous and exogenous damaging agents. The BER protects the genome of the organism from various DNA damages caused by oxidation, alkylation and deamination [39]. BER system is a highly conserved system employed from bacteria to humans responsible for the removal of a large number of endogenous DNA damages which include deamination, depurination and alkylation [39]. There is unequivocal understating that any defect in BER pathway may ultimately lead to cancer. On other face, manipulation or alterations to BER mechanisms may prove to act as tools to survive during genotoxic threat.
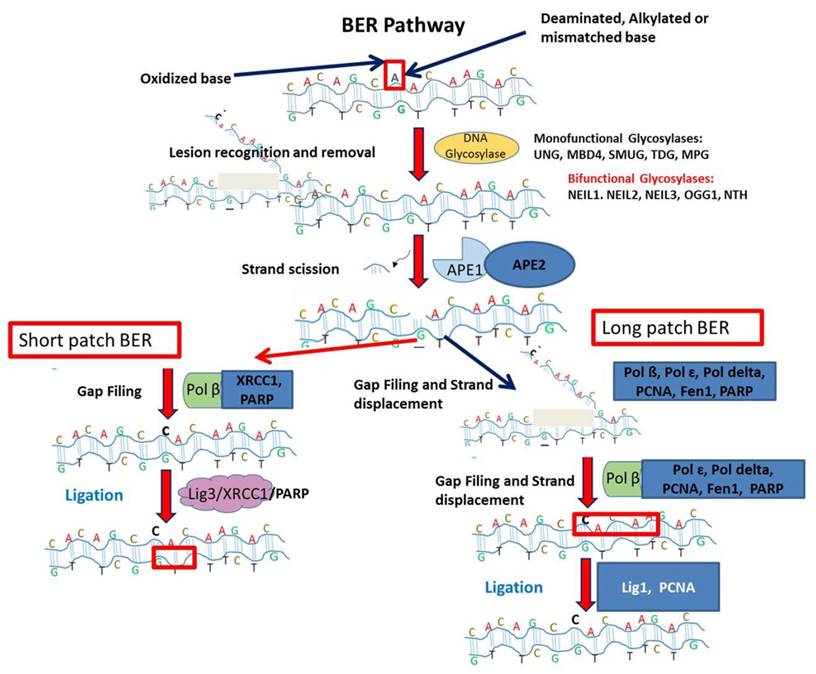
The BER pathway is a complex interplay of many enzymes. The detailed schematic representation is given in Figure 1 [40, 41, 42]. The first enzyme in BER pathway is the DNA glycosylase and specific for different types of DNA damage [41]. Different lesions are treated by lesion specific DNA glycosylases. Presently, eleven different DNA glycosylases are known. The major role of these glycosylases is to excise the bond between deoxyribose and incorrectly placed nitrogenous base creating apurinic or apyridiminic site [42]. The mode of action of DNA glycosylases are monofunctional or bifunctional. The monofunctional glycosylases use water as nucleophile to remove the damaged base whereas later uses amine moiety for nucleophilic attack. The monofunctional glycosylases is a Uracil DNA Glycosylase (UDG) superfamily which has further four lesion specific DNA glycosylases like uracil DNA N-glycosylase (UNG), thymine DNA glycosylase (TDG), single-strand-selective mono-functional uracil-DNA glycosylase 1 (SMUG1) and methyl-CpG-binding domain 4 (MBD4). The DNA duplex due to reactive oxygen species is acted by DNA glycosylases as MutY homolog (MYH), 8-oxoguanine glycosylase 1 (OGG1), endonuclease three homolog 1 (NTH1) and Nei endonuclease VIII-like 1, 2, and 3 (NEIL1, NEIL2, and NEIL3) [41]. BER is an evolutionary conserved process to maintain the genomic integrity by removal of damage bases generated by genotoxicants.
Next, AP Endonucleases are the next enzyme carrying the in BER pathway. The enzyme APE1 is described as the major AP-endonuclease in mammalian cells and essential for the survival [41]. The role of this enzyme is to generate the 5'-sugar phosphate group (dRP) and 3'-OH ends by incising the phosphodiester bond of DNA in the presence of Mg2+ ions [41,43]. The major class of AP endonuclease is APE 1 and referred as HAP 1 and Alex [42]. The APE 1 do not only functions for endonuclease activity but also it serves redox functions for activation of several transcription factors. Other than these functions, it possess proof reading role and catalyzes the removal of 3'-blocking agents generated by bifunctional DNA glycosylases [41].Several studies are linked with the differential expression profiles of BER enzymes such as (redox factor 1/apurinic apyrimidinic endonuclease 1 (REF1/APEX1), NTH endonuclease III-like 1 (NTHL1), 8-oxoguanine DNA glycosylase (OGG1), PARP-1 and scaffold protein XRCC1 (X-ray repair complementing defective repair in Chinese hamster cells 1) in familial and sporadic breast cancer. Based on expression study, there are reports on the upregulation of APEX1/REF1 in familial BRCA-wild-type and sporadic breast cancer cases indicating potential therapeutic role.
The process of repair is further followed by DNA polymerase and DNA ligase (for nick sealing) [40]. DNA Polymerases are defined as essential part of BER pathway to lyse the blocking 5'- dRP site and further synthesizes the new nucleotides at 3' end of the nick [44]. DNA polymerase β is noted as the major polymerase type in BER. This enzyme is reported to contain two catalytic domains: one is N terminal domain for 5' dRP lyase and C- terminal as DNA binding domain [45]. The BER Pathway are mentioned as two types as nucleotides added to the abasic site i.e. short patch repair pathway (addition of 1-2 nucleotides) and long patch repair pathway (addition of 2-8 nucleotides) [44,46]. During BER pathway, different polymerases are used for both pathways. DNA polymerase β is used in short patch repair pathway. But in long path BER, 5'-dRP is resistant to cleavage by pol β. Therefore, switch from pol β to pol δ/ϵ takes place. But first nucleotide is synthesized by pol β. Then, 2-8 nucleotides are synthesized by pol δ/ϵ thereby generating a flap like structure which is removed by FEN 1 [45]. Other than these DNA polymerases, there is report on another polymerase as pol λ which plays backup role in BER. It is 32% homologous to pol β and plays role in repairing oxidative lesions and intrinsic 5'- dRP lyase activity [47].
After the synthesis of new nucleotides, DNA ligases perform the sealing process. DNA ligase encoded by gene LIG 3 generates a covalent phosphodiester bond between 3'- OH and 5' -PO4 using ATP/NAD+ as the energy source. The long patch BER uses DNA ligase 1 while short patch BER uses Ligase III α/ XRCC1 complex. The interaction of proteins of this complex is thought to mediate by BRCT domains of both (L3BRCT and X1BRCT). But the complex that mediates their interaction is still unknown. Several structural models are suggested to explain their tetrameric quaternary complex. It is also suggested that L3BRCT and X1BRCT connected by some N-terminal linker region [48]. Casein kinase 2 (CK2) keeps XRCC1 phosphorylated owing to the stability of XRCC-Lig III complex [49]. The DNA Ligase III shows damage sensing ability attributed by its Zn domain at N- terminus homologous to PARP1.
This illustration depicts Base excision repair (BER) pathway. DNA glycosylase recognizes damaged lesion. Further, AP endonuclease cleaves damaged base. Followed by gap filling step in short patch BER is executed by Pol ß, X-ray repair cross-complementing protein 1 (XRCC1) and PARP. For Long patch BER, the gap filling and strand displacement are performed by Pol ß, Pol ɛ, Pol delta, Proliferating cell nuclear antigen (PCNA), Flap endonuclease 1 (Fen1) and poly ADP-ribose polymerase (PARP). The last ligation step is carried out by DNA ligase III (Lig3) in short patch BER. In long patch BER, the ligation step is completed by DNA ligase I (Lig1).
In the pool of several key DNA repair response proteins, Poly (ADP- Ribose) Polymerases (PARP) are described as a superfamily of seventeen proteins. They play a key role in cellular processes such as telomere homeostasis, chromatin dynamics, cell differentiation and cell death [50]. The PARPs are reported as essential proteins required in BER. They play crucial role in sensing DNA damage and promoting repair [51]. The enzyme PARP is extensively referred for its role in Poly(ADP-ribosylation) (PARylation process as a post-translational modification of proteins engaged in DNA damage repair. This enzyme recognizes the damage and bind to damaged site by cleaving NAD+ and addition of ADP- ribose to form long and branched chains of Poly (ADP-ribose) [PAR]. The PAR is covalently bonded to acceptor proteins forming polymers around the damage thereby recruiting other essential proteins of BER. The detailed biochemical and functional characterization of the DNA-dependent PARP family members are explained as three members as PARP 1, PARP 2 and PARP 3. Among these members, PARP1 and PARP2 are accentuated for their participation in the resolution of single-strand breaks as part of the BER/SSBR process [52]. The PARP 1 is reported to possess three domains as NH2 terminal DNA binding domain having 3 Zn fingers (2nd Zn finger with strongest affinity to DNA break), automodification domain and C-terminal catalysis domain [53]. The hyperactivation of PARP 1 leads to depletion of NAD+ reserves thus leading to necrosis or apoptosis of cell. This property of PARP is used to attack cancer cells. Some PARP inhibitors bind to catalytic domain inhibiting auto modification thereby removing enzyme from site of DNA damage [52]. However, PARylation is also reported to contribute to the repair of double-strand breaks [54, 55].
BER and Epigenetic at Crossroad in Breast Carcinoma
Several types of cancer including breast carcinoma are depicted as combined landscape created by both genetic and epigenetic events [56]. In recent view, the epigenetic factors are suggested to play critical role in the DNA damage response regulated by multiple elements. Epigenetic landscape is maintained by the concerted efforts from epigenome molecular payers linked with DDR in normal and carcinoma cells. Further, therapeutic approaches are considered to target epigenetic machinery components [57, 58]. By dissecting out BER response and epigenetic landscape cross talk, there are appreciable suggestions that BER pathway influenced by chromatin disruption and remodeling [59]. The DNA methylation is known as a stable epigenetic modification. In recent, the existing role of DNA methyl transferase transferases 3A and 3B (DNMT-3A and 3B) is considered to carry out DNA methylation and also being extended to demonstrate active DNA demethylation. Further, BER pathway has been suggested to facilitate demethylation C-5 methyl cytosine (5mC) by DNMT-3A and -3B might be in the absence of S-adenosyl methionine (SAM) [60].
There is consensus that epigenetic landscape in cancer cells are highly aberrant and suggested to act as the promoter for the malignancy. In recent, data accumulated from genomic approaches point out that several miRNAs are deregulated in breast cancer [61, 62]. Besides, a player involved in the miRNA maturation as endoribonuclease Dicer is also reported to be down regulated in breast carcinoma [61, 62]. The Gadd45a (growth arrest and DNA-damage-inducible protein 45 alpha) is reported as nuclear protein to maintain genomic stability, DNA repair, cell growth retardation and DNA demethylation. Their findings substantiate that active demethylation may be facilitated via DNA repair response mechanisms, where Gadd45a interacts with DNA repair endonuclease XPG [63].Recently, evidence indicates that in breast carcinoma, histone deacetylase (HDAC) inhibitors bring rapid alterations in the expression of MYC controlled miRNAs, including let-7 family [64].There is reports on the implications of two repair enzymes, thymine DNA glycosylase (TDG) and methyl-CpG binding domain 4 (MBD4) against the mutagenic role of methyl-cytosines in mammalian system. Further, authors have established about the convergence between DNA repair and re-methylation at the methyl-cytosine deamination chromosomal loci [65].
BER and microRNA Regulation in Breast Carcinoma
Recently, microRNAs (miRNAs) defined as non-coding regulatory players are acknowledged for their decisive role in numerous normal biological processes besides cancer progression and development [66, 67]. In the perspectives of miRNAs action in cancer pathophysiology, miRNAs are suggested as potentials biomarkers for diagnostic and prognostic approaches in cancer treatment. There is view that therapeutic approaches in the form of bringing down or elevating levels of specific miRNAs by designing anti-sense oligonucleotides or pre-miRNAs may be useful for the cancer therapy [67, 68]. In recent, obstacles in the form of highly resisting cancer stem cells are reported in several carcinomas. Therefore, search of selective agents such as natural compounds curcumin or epigallocatechin-3-gallate (EGCG) are reported for their role to modulate miRNAs in such type of cells [69].
There is growing evidence to support that approximately 50% of miRNA-encoding genes are situated in the chromosomal loci associated with cancer pathophysiology [70, 71, 72]. These diverse set of miRNAs sometimes referred either as oncogenic miRNAs (oncomiRNAs) in case upregulated or tumor suppressors of repressed in a specific type of cancer. Among reported cancer related miRNAs, miRNA-15a and miRNA16-1 are suggested to play as the tumor suppressor's role in solid tumors including ovarian and breast cancer [72, 73]. In recent, investigations are reported that miRNAs screening could be an effective means of detecting down regulation of gene products associated with DNA damage response and breast cancer phenotypes [74].
Recently, MiR-155 is reported to act asoncomiRNAs, which is highly elevated in breast cancer and linked to the estrogen receptor (ER) and progesterone receptor (PR) level. The author Zhang and colleague report that that tumor protein 53-induced nuclear protein 1(TP53INP1) could be potential targets of miRNA-155 in case of ERalpha (+) MCF-7 cancer cell lines. Furthermore, their findings confirm that overexpression of miRNA-155 can block TP53INP1in MCF-7 cells and possibly help in cellular proliferation and blocking apoptosis. In addition, findings indicated that miR-155 can obstruct the cell cycle and apoptosis-related gene caspase-3, -8, -9, and p21 expression [75]. There are finding to support that overexpression of miR-21 in breast cancer can lead to invasiveness and lung metastasis [76, 77].
Therapeutic Intervention of Drugs/Inhibitors against BER in Breast Carcinoma
BER is highlighted as the main process to maintain the integrity of the genome inflicted due to genotoxic agents [10-12,14]. The primary step in repairing a single base damage in DNA is glycosylase activities. Abasic sites are generated following glycosylase removal of a damaged base followed by the action of APE1/Ref-1 enzyme. Recently, Donley et al (2015) reported about the potent and specific inhibitors to OGG1 DNA glycosylase and most of them classified as hydrazides or acyl hydrazones. There are suggestions that APE1 is a better therapeutic target [78]. Aberrant BER responses contribute to malignant transformation. However, an inter-individual variation in DNA repair capacity plays a key role in modifying breast cancer risk and possible success or failure of genotoxic approaches [79]. There are reports on emerging evidences about the aberrant expression of several BER proteins and among those adenomatous polyposis coli (APC) and Flap endonuclease 1 (Fen1) expression in breast tumors [80]. It is considered that increasing the concentration of APC may enhance the efficacy of the cytotoxic drugs that will enhance cell death by producing DNA damage. In actual pathway, APC interacts with and blocks Fen1 activity in Pol-β-directed LP-BER, and abrogation of LP-BER linked with transformation of normal breast epithelial cells [79]. The APE is explained as one of the main enzymes in the BER pathway and creates nearly all of the abasic site cleavage activity. The APE1 expression is distinct in variety of cancer and a high APE1 expression associated with poor result towards chemo-radiotherapy [81]. Many APE1 chemical inhibitors are isolated among as CRT0044876 a potent and selective APE1 inhibitor. In silico modeling studies suggest that CRT0044876 binds to the active site of APE1 [82]. Methoxyamine is revealed as a small molecule that specifically inhibits BER. Methoxyamine inhibits the repair of AP sites by binding to and modifying the AP site, rather than directly inhibiting the enzyme APE1 [83]. Inhibition of AP-endonuclease (APE) lead to increased sensitivity of cancer cells to alkylating chemotherapeutics [84]. When AP-sites are oxidized or reduced, they become resistant to elimination and cannot be excised by the dRP lyase activity of pol [85]. Many genes play a key role in BER like X-ray repair cross-complementing group 1 (XRCC1), OGG1 gene, PARP-1 and APE1 genes [55]. The XRCC1 interacts with several BER proteins such as DNA polymerase β, APE1, OGG1, PARP-1 and LIGIII [86]. X‐ray Repair Cross Complementing 1 (XRCC1) is thought to function as a scaffolding protein in BER pathway [87]. XRCC1 is a multidomain protein that interacts with nicked DNA and participates with at least three enzymes PARP, DNA ligase III, DNA polymerase [88]. XRCC3 is a member of the Rad51 DNA repair gene family; it functions in the HRR pathway for repairing double-strand breaks, which plays important roles in maintaining genome stability [89, 90]. XRCC1 protein make complex with the PARP-1 enabling their recruitment in damage site [91]. In view of combinatorial drug therapy prospects, authors have demonstrated that pan-histone deacetylase inhibitor (HDI) treatment in combination with PARP inhibitor against TNBC cells resulted into enhanced lethality [92].
In recent, efforts are available to delineate alkylating agent responsiveness in TNBC to interfere the level of hPMC2 leading to diminished cell survival [93]. The combinatorial effect of Doxorubicin (Dox) and Methylamine (MX) on MDA-MB-231 depicts a sensitizing effect. In this report, Dox helps in inducing an oxidative DNA damage whereas MX acts an apurinic/apyrimidinic endonuclease 1 (APE1) inhibitor [94]. Recently, combinatorial drugs/inhibitors treatment regiments are described involving PARP and PI3K pathway inhibitors. The human breast cancer cell lines demonstrated heightened cell death and apoptosis in case of combined use of PARP and PI3K inhibitors [5, 20, 95]. Recently, olaparib (Lynparza), PARP inhibitor are directed towards cancer types with debilitating DNA repair pathways as BRCA1 and BRCA2 mutation [10]. The cellular effects of PARP inhibitors are reported to vary according to the cellular environment. In particular, the presence and nature of DNA damage and/or metabolic stress leave impact on the consequences of PARP inhibition [96]. The inhibitors of PARP-1 (a nuclear enzyme involved in the signaling of DNA damage and BER) in conjunction with radiation or cytotoxic drug as topoisomerase (TOPO) type I or II inhibitors can induce severe genomic instability [97].
Conclusion
In conclusion, current regimens of genotoxic drug and radiation therapy based treatment are successful in some cases. At the same time, failures widely witnessed in breast carcinoma. It is true that several carcinomas including breast cancer are blessed with modulated dedicated pool of DDR pathways including BER pathway. The efforts to use the knowledge about BER proteins and their interference in breast carcinoma will offer opportunities for cocktails of precise genotoxic drugs/inhibitors.
Acknowledgements
We would like to thank the Cancer and Translational Research Lab for their constant support and motivation and our host institute Dr. D. Y Patil Biotechnology & Bioinformatics Institute, Dr. D. Y Patil Vidyapeeth, Pune for providing resources. The authors acknowledge the financial support from DST, SERB, New Delhi and DPU Pune. We would like to convey our sincere apologies to the authors whose citations are included indirectly and also to the ones which are not cited.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Carey LA, Perou CM, Livasy CA. et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA. 2006;295(21):2492-2502
2. Rakha EA, Reis-Filho JS, Ellis IO. Basal-like breast cancer: a critical review. Journal of ClinOncol. 2008;26(15):2568-2581
3. Badve S, Dabbs D, Schnitt S. et al. Basal-like and triple-negative breast cancers: a critical review with an emphasis on the implications for pathologists and oncologists. Mod Pathol. 2011;24:157-167
4. Bogdanova N, Helbig S, Dörk T. Hereditary breast cancer: ever more pieces to the polygenic puzzle. Hered Cancer ClinPract. 2013;11(1):12
5. Kimbung S, Loman N, Hedenfalk I. Clinical and molecular complexity of breast cancer metastases. Semin Cancer Biol. 2015;35:85-95
6. Braunstein LZ, Taghian AG. Molecular Phenotype, Multigene Assays, and the Locoregional Management of Breast Cancer. SeminRadiatOncol. 2016;26(1):9-16
7. Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411(6835):366-74
8. Lindahl T, Wood RD. Quality control by DNA repair. Science. 1999;286(5446):1897-905
9. Holohan C, Van Schaeybroeck S, Longley DB, et al.PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13(10):714-726
10. O'Connor MJ. Targeting the DNA Damage Response in Cancer. Mol Cell. 2015;60(4):547-560
11. Khanna A. DNA damage in cancer therapeutics: a boon or a curse? Cancer Res. 2015;5(11):2133-8
12. Gao D, Herman JG, Guo M. The clinical value of aberrant epigenetic changes of DNA damage repair genes in human cancer. Oncotarget. 2016 doi: 10.18632/oncotarget.7949. [Epub ahead of print]
13. Telli ML, Sledge GW. The future of breast cancer systemic therapy: the next 10 years. J Mol Med (Berl). 2015;93(2):119-125
14. Gavande NS, VanderVere-Carozza PS, Hinshaw HD. et al. DNA repair targeted therapy: The past or future of cancer treatment? PharmacolTher. 2016 doi: 10.1016/j.pharmthera.2016.02.003
15. Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461(7267):1071-8
16. Bauer NC, Corbett AH, Doetsch PW. The current state of eukaryotic DNA base damage and repair. Nucleic Acids Res. 2015;43(21):10083-101
17. Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic targets. Nature Reviews Cancer. 2012;12(12):801-817
18. Shaheen M, Allen C, Nickoloff JA. et al. Synthetic lethality: exploiting the addiction of cancer to DNA repair. Blood. 2011;117(23):6074-82
19. Liu Y, Li Y, Lu X. Regulators in the DNA damage response. Arch BiochemBiophys. 2016;594:18-25
20. Puigvert JC, Sanjiv K, Helleday T. Targeting DNA repair, DNA metabolism and replication stress as anti-cancer strategies. FEBS J. 2016;283(2):232-245
21. Martinez GR, Loureiro APM, Marques SA. et al. Oxidative and alkylating damage in DNA. Mutat Res. 2003;544(2):115-127
22. De Bont R, van Larebeke N. Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis. 2004May;19(3):169-85
23. Chen X, Zhong S, Zhu X. et al. Rational design of human DNA ligase inhibitors that target cellular DNA replication and repair. Cancer Res. 2008;68(9):3169-77
24. Singh DK, Krishna S, Chandra S. et al. Human DNA ligases: a comprehensive new look for cancer therapy. Medical Res Rev. 2013;34(3):567-595
25. Gruver AM, Portier BP, Tubbs RR. Molecular pathology of breast cancer: the journey from traditional practice toward embracing the complexity of a molecular classification. Arch Pathol Lab Med. 2011;135(5):544-57
26. Goncalves R, Warner WA, Luo J. et al. New concepts in breast cancer genomics and genetics. Breast Cancer Res. 2014;16(5):460
27. Aleskandarany M, Caracappa D, Nolan CC. et al. DNA damage response markers are differentially expressed in BRCA-mutated breast cancers. Breast Cancer Res Treat. 2015;150(1):81-90
28. Zhang Y, Newcomb PA, Egan KM. et al. Genetic polymorphisms in base-excision repair pathway genes and risk of breast cancer. Cancer Epidemiol Biomarkers Prev. 2006;15(2):353-358
29. Collignon J, Lousberg L, Schroeder H. et al. Triple-negative breast cancer: treatment challenges and solutions. Breast Cancer (Dove Med Press). 2016;8:93-107
30. Kleibl Z, Kristensen VN. Women at high risk of breast cancer: Molecular characteristics, clinical presentation and management. The Breast. 2016;28:136-144
31. Menghi F, Inaki K, Woo X. et al. The tandem duplicator phenotype as a distinct genomic configuration in cancer. Proc Natl AcadSci U S A. 2016;113(17):E2373-82
32. Hydbring P, Sanda T, Stefano J. et al. The requirement for cyclin D function in tumor maintenance. Cancer Cell. 2012;22(4):438-51
33. Id Said, Kim H. et al. Super-transactivation TP53 Variant in the Germline of a Family with Li-Fraumeni Syndrome. Human Mut. 2016;37(9):889-92
34. Schiewer MJ, Knudsen KE. Linking DNA Damage and Hormone Signaling Pathways in Cancer. Trends EndocrinolMetab. 2016;27(4):216-25
35. Wood RD, Mitchell M, Sgouros J. et al. Human DNA repair genes. Science. 2001;291(5507):1284-9
36. Akbari M, Peña-Diaz J, Andersen S. et al. Extracts of proliferating and non-proliferating human cells display different base excision pathways and repair fidelity. DNA Repair. 2009;8(7):834-843
37. Hosoya N, Miyagawa K. Targeting DNA damage response in cancer therapy. Cancer Science. 2014;105(4):370-388
38. London RE. The structural basis of XRCC1-mediated DNA repair. DNA Repair (Amst). 2015;30:90-103
39. Wallace SS. Base excision repair: a critical player in many games. DNA Repair (Amst). 2014;19:14-26
40. Hegde ML, Hazra TK, Mitra S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 2008;18(1):27-47
41. Kim YJ, M Wilson III D. Overview of base excision repair biochemistry. CurrMolPharmacol. 2012;5(1):3-13
42. Krokan HE, Bjørås M. Base excision repair. Cold Spring Harbor Perspectives in Biology. 2013;5(4):12583
43. Sukhanova MV, Khodyreva SN, Lebedeva NA. et al. Human base excision repair enzymes apurinic/apyrimidinic endonuclease1 (APE1), DNA polymerase β and poly (ADP-ribose) polymerase 1: interplay between strand-displacement DNA synthesis and proofreading exonuclease activity. Nucleic Acid Res. 2005;33(4):1222-1229
44. Dianov GL, Hübscher U. Mammalian base excision repair: the forgotten archangel. Nucleic Acids Res. 2013;41(6):3483-90
45. Nicolay HN, Helleday T, A Sharma RA. Biological relevance of DNA polymerase beta and translesion synthesis polymerases to cancer and its treatment. CurrMolPharmacol. 2012;5(1):54-67
46. Prasad R, Williams JG, Hou EW. et al. Pol β associated complex and base excision repair factors in mouse fibroblasts. Nucleic Acid Research. 2012;40(22):11571-11582
47. Bebenek K, Pedersen LC, Kunkel TA. Structure-function studies of DNA polymerase λ. Biochemistry. 2014;53(17):2781-2792
48. Cuneo MJ, Gabel SA, Krahn JM. et al. The structural basis for partitioning of the XRCC1/DNA ligase III-α BRCT-mediated dimer complexes. Nucleic acids research. 2011;39(17):7816-27
49. Parsons JL, Dianova II, Finch D. et al. XRCC1 phosphorylation by CK2 is required for its stability and efficient DNA repair. DNA Repair. 2010;9(7):835-84
50. De Vos M, Schreiber V, Dantzer F. The diverse roles and clinical relevance of PARPs in DNA damage repair: current state of the art. BiochemPharmacol. 2012;84(2):137-16
51. Langelier MF, Planck JL, Roy S. et al. Crystal structures of poly (ADP-ribose) polymerase-1 (PARP-1) zinc fingers bound to DNA structural and functional insights into DNA-dependent PARP-1 activity. J Biol Chem. 2011;286(12):10690-10701
52. Chen A. PARP inhibitors: its role in treatment of cancer. Chinese Journal of Cancer. 2011;30(7):463
53. Krishnakumar R, Kraus WL. The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol Cell. 2010;39(1):8-24
54. Beck C, Robert I, Reina-San-Martin B. et al. Poly(ADP-ribose) polymerases in double-strand break repair: focus on PARP1, PARP2 and PARP3. Exp Cell Res. 2014;329(1):18-25
55. Patrono C, Sterpone S, Testa A. et al. Polymorphisms in base excision repair genes: Breast cancer risk and individual radiosensitivity. World J ClinOncol. 2014;5(5):874-82
56. Lv JF, Hu L, Zhuo W. et al. Epigenetic alternations and cancer chemotherapy response. Cancer ChemotherPharmacol. 2016;77(4):673-84
57. Dabin J, Fortuny A, Polo SE. Epigenome Maintenance in Response to DNA Damage. Mol Cell. 2016;62(5):712-27
58. Montenegro MF, González-Guerrero R, Sánchez-Del-Campo L. et al. Targeting the epigenetics of the DNA damage response in breast cancer. Cell Death Dis. 2016;7:e2180. doi: 10.1038/cddis.2016.85
59. Odell ID, Wallace SS, Pederson DS. Rules of engagement for base excision repair in chromatin. J Cell Physiol. 2013;8(2):258-66
60. van der Wijst MG, Venkiteswaran M, Chen H. et al. Local chromatin microenvironment determines DNMT activity: from DNA methyltransferase to DNA demethylase or DNA dehydroxymethylase. Epigenetics. 2015;10(8):671-676
61. Grelier G, Voirin N, Ay AS. et al. Prognostic value of Dicer expression in human breast cancers and association with the mesenchymal phenotype. Br J Cancer. 2009;101:673-683
62. Veeck J, Esteller M. Breast cancer epigenetics: from DNA methylation to microRNAs. J Mammary Gland Biol Neoplasia. 2010;15(1):5-17
63. Barreto G, Schäfer A, Marhold J. et al. Gadd45a promotes epigenetic gene activation by repair-mediated DNA demethylation. Nature. 2007;445(7128):671-675
64. Adams CM, Eischen CM. Histone deacetylase inhibition reveals a tumor-suppressive function of MYC-regulated miRNA in breast and lung carcinoma. Cell Death Differ. 2016;23(8):1312-1321
65. Li YQ, Zhou PZ, Zheng XD. et al. Association of Dnmt3a and thymine DNA glycosylase links DNA methylation with base-excision repair. Nucleic Acids Res. 2007;35(2):390-400
66. Ghildiyal M, Zamore PD. 2009. Small silencing RNAs: an expanding universe. Nat Rev Genet. 2016;10:94-108
67. Sarkar FH, Li Y, Wang Z. et al. Implication of microRNAs in drug resistance for designing novel cancer therapy. Drug Resist Updat. 2010;13(3):57-66
68. Harquail J, Benzina S, Robichaud GA. MicroRNA and Breast cancer malignancy: An overview of miRNA-regulated cancer processes leading to metastasis. Cancer Biomarkers. 2012;11(6):269-280
69. Wang Z, Li Y, Ahmad A. et al. Targeting miRNAs involved in cancer stem cell and EMT regulation: An emerging concept in overcoming drug resistance. Drug Resist Updat. 2010;13(4):109-118
70. Esquela-Kerscher A, Johnson SM, Bai L. et al. Oncomirs — microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:59-69
71. Kent OA, Mendell JT. A small piece in the cancer puzzle: microRNAs as tumor suppressors and oncogenes. Oncogene. 2006;25:6188-96
72. Nikitina EG, Urazova LN, Stegny VN. MicroRNAs and human cancer. ExpOncol. 2012;34(1):2-8
73. Zhang L, Huang J, Yang N. MicroRNAs exhibit high frequency genomic alterations in human cancer. Proc Natl AcadSci USA. 2006;103:9136-41
74. Bottai G, Pasculli B, Calin GA. et al. Targeting the microRNA-regulating DNA damage/repair pathways in cancer. Expert OpinBiolTher. 2014;14(11):1667-1683
75. Zhang CM, Zhao J, Deng HY. MiR-155 promotes proliferation of human breast cancer MCF-7 cells through targeting tumor protein 53-induced nuclear protein 1. J Biomed Sci. 2013;20:79
76. Iorio MV, Ferracin M, Liu CG. et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65:7065-7070
77. Zhu S, Wu H, Wu F. et al. MicroRNA-21 targets tumor suppressor genes in invasion and metastasis. Cell Res. 2008;18:350-359
78. Kelley MR, Fishel ML. DNA repair proteins as molecular targets for cancer therapeutics. Anticancer Agents Med Chem. 2008;8(4):417-425
79. Narayan S, Jaiswal AS, Law BK. et al. Interaction between APC and Fen1 during breast carcinogenesis. DNA Repair (Amst). 2016;41:54-62
80. Jaiswal AS, Armas ML, Izumi T. et al. Adenomatous polyposis coli interacts with flap endonuclease 1 to block its nuclear entry and function. Neoplasia. 2012;14(6):495-50
81. Luo M, Kelley MR. Inhibition of the human apurinic/apyrimidinic endonuclease (APE1) repair activity and sensitization of breast cancer cells to DNA alkylating agents with lucanthone. Anticancer Res. 2004;24(4):2127-2134
82. Madhusudan S, Smart F, Shrimpton P. et al. Isolation of a small molecule inhibitor of DNA base excision repair. Nucleic Acids Res. 2005;33(15):4711-4724
83. Tang JB, Goellner EM, Wang XH. et al. Bioenergetic metabolites regulate base excision repair-dependent cell death in response to DNA damage. Mol Cancer Res. 2010;8(1):67-79
84. Adhikari S, Choudhury S, Mitra PS. et al. Targeting base excision repair for chemosensitization. Anticancer Agents Med Chem. 2008;8(4):351-357
85. Kundu C, Balusu R, Jaiswal AS. et al. Cigarette smoke condensate-induced level of adenomatous polyposis coli blocks long-patch base excision repair in breast epithelial cells. Oncogene. 2007;26(10):1428-1438
86. Sterpone S, Cozzi R. Influence of XRCC1 Genetic Polymorphisms on Ionizing Radiation-Induced DNA Damage and Repair. Journal of Nucleic Acids. 2010 doi: 10.4061/2010/780369
87. Chacko P, Rajan B, Joseph T. et al. Polymorphisms in DNA repair gene XRCC1 and increased genetic susceptibility to breast cancer. Breast cancer research and treatment. 2005;89(1):15-21
88. Kim SU, Park SK, Yoo KY. et al. XRCC1 genetic polymorphism and breast cancer risk. Pharmacogenetics and Genomics. 2002;12(4):335-338
89. Smith TR. et al. Genetic polymorphisms of multiple DNA repair pathways impact age at diagnosis and TP53 mutations in breast cancer. Carcinogenesis. 2011;32(9):1354-60
90. Sultana et al. Targeting XRCC1 deficiency in breast cancer for personalized therapy. Cancer Res. 2013;73(5):1621-34
91. Horton JK. Wilson SH. Predicting enhanced cell killing through PARP inhibition. Molecular Cancer Res. 2013;11(1):13-18
92. Ha K, Fiskus W, Choi DS. et al. Histone deacetylase inhibitor treatment induces 'BRCAness' and synergistic lethality with PARP inhibitor and cisplatin against human triple negative breast cancer cells. Oncotarget. 2014;5(14):5637-50
93. Krishnamurthy N, Liu L, Xiong X. et al. Downregulation of hPMC2 imparts chemotherapeutic sensitivity to alkylating agents in breast cancer cells. Cancer BiolTher. 2015;16(4):518-27
94. Guerreiro PS, Fernandes AS, Costa JG. et al. Differential effects of methoxyamine on doxorubicin cytotoxicity and genotoxicity in MDA-MB-231 human breast cancer cells. Mutat Res. 2013;757(2):140-7
95. Livraghi L, Garber JE. PARP inhibitors in the management of breast cancer: current data and future prospects. BMC Med. 2015;13:188
96. Chalmers AJ. The potential role and application of PARP inhibitors in cancer treatment. British Medical Bulletin. 2009;89(1):23-40
97. Węsierska-Gądek J1, Zulehner N, Ferk F. et al. PARP inhibition potentiates the cytotoxic activity of C-1305, a selective inhibitor of topoisomerase II, in human BRCA1-positive breast cancer cells. BiochemPharmacol. 2012;84(10):1318-1331
Author contact
![]() Corresponding author: Dr. Nilesh Kumar Sharma, Cancer and Translational Research Lab, Dr. D.Y Patil Biotechnology & Bioinformatics Institute, Padmashree Dr. D. Y. Patil University, Pune, Maharashtra, India, 411033. Email: nilesh.sharmaedu.in Telephone: +91-02067919400 Fax: +91-02067919444.
Corresponding author: Dr. Nilesh Kumar Sharma, Cancer and Translational Research Lab, Dr. D.Y Patil Biotechnology & Bioinformatics Institute, Padmashree Dr. D. Y. Patil University, Pune, Maharashtra, India, 411033. Email: nilesh.sharmaedu.in Telephone: +91-02067919400 Fax: +91-02067919444.