Oncomedicine 2017; 2:88-92. doi:10.7150/oncm.19362 This volume Cite
Research Paper
Coexistence of Two Novel Mutations in CDKN2A and PMS1 Genes in a Single Patient Identifies a New and Severe Cancer Predisposition Syndrome
Marco Cassone1 ![]() , Leila Baghernajad Salehi1, Gianluca Tedaldi2, Michela Tebaldi2, Valentina Zampiga2, Michela Biancolella1, Barbara Testa1, Daniele Calistri2, Gerarda Mastrogiorgio1, Silvia Lanciotti1, Maria Rosaria D'Apice3, Giuseppe Novelli1, Federica Sangiuolo1
, Leila Baghernajad Salehi1, Gianluca Tedaldi2, Michela Tebaldi2, Valentina Zampiga2, Michela Biancolella1, Barbara Testa1, Daniele Calistri2, Gerarda Mastrogiorgio1, Silvia Lanciotti1, Maria Rosaria D'Apice3, Giuseppe Novelli1, Federica Sangiuolo1
1. Department of biomedicine and prevention, University Tor Vergata Rome, Rome, Italy;
2. Biosciences Laboratory, Istituto Scientifico Romagnolo per lo Studio e la Cura dei Tumori (IRST) IRCCS, Meldola (Forlì-Cesena), Italy;
3. Medical Genetics Laboratory, Policlinico Tor Vergata, Rome, Italy.
Received 2017-1-26; Accepted 2017-4-6; Published 2017-5-4
Abstract
Introduction: Up to 10% of cancers occur through the inherited mutation of a group of genes called cancer predisposition genes [1]. Carriers of monoallelic mutations of these genes are associated with an increased susceptibility to cancer. Autosomal dominant cancer predisposition genes for common cancers have been well recognized for over decade. Each newly identified cancer predisposition gene has been associated with a distinctive autosomal dominant or recessive cancer syndrome [2]. The CDKN2A is the major known melanoma susceptibility gene and has been associated with Familial atypical multiple mole melanoma syndrome (FAMMM #155601), while PMS1 gene is involved in mismatch repair (MMR) process and its mutations have been associated with Lynch syndrome (LS) (HNPCC for hereditary non polyposis colorectal cancers) (OMIM #120435) in few cases [3].
Materials and methods: Genetic counseling, molecular analysis of 94 genes by using ''IlluminaTruSight Cancer'' panel.
Results: We have detected in a patient with a novel frameshift mutation within PMS1 gene, PMS1c.1139dupA p.Y380_S381delinsX, and in the same time a novel mutation within CDKN2a gene, CDKN2a c.58delG p.V20X. In this way, the coexistence of two germline mutations in two different genes, already associated to cancer predisposition syndrome, such as LS and FAMMM have been described.
Discussion: Our data frame this case as a new and severe cancer predisposition syndrome. An appropriate genetic counseling surely represents the key step for a correct test choice in panel genes era.
Keywords: PMS1, CDKN2A, Lynch syndrome, cancer predisposition syndrome, familial melanoma.
Introduction
Up to 10% of cancers occur through the inherited mutation of a group of genes called 'cancer predisposition genes. Individuals who carry a mutant allele of these genes have an increased susceptibility to cancer.
A growing number of cancer susceptibility genes are being identified and the physiopathology of germline mutation-based cancer development is also being elucidated with accumulating clinical and molecular data. New technologies for DNA sequencing allow us to analyze simultaneously multiple genes assembled in specific panels and associated with cancer predisposition, offering a useful tool in the common clinical practice of genetic counseling. The characterization of a genetic cause of cancer plays an extremely important role for the treatment, which may differs from the treatment of sporadic disease, and for periodic surveillance.
The first description of inherited cancers was established in "family G" pedigree with an impressive aggregation of colon and gynecological cancers in family members throughout four generations [3]. The family was reinvestigated from Lynch and Krush in 1971 and the predisposition appeared to be inherited in an autosomal dominant manner [4].
The syndrome was named "Lynch Syndrome" (LS) or HNPCC for hereditary non polyposis colorectal cancers (OMIM #120435), one of the most common cancer syndrome, accounting for 1-3% of unselected colorectal carcinomas. Lynch syndrome is an autosomal dominant condition caused by heterozygous germline mutations in the DNA mismatch repair genes, the PMS1 is recognized as (MMR) genes and its mutations were associated with LS in a few cases PMS1 [5-6]. Rare cases have been reported of an inherited bi-allelic deficiency of MMR genes, associated with multiple café-au-lait spots, early onset CNS tumors, hematological malignancies, and early onset gastrointestinal neoplasia [7-8].
There are also a number of less frequent syndromes with distinctive cancer spectrums. Among them, Familial Atypical Multiple Mole Melanoma (FAMMM#155601) and hereditary melanoma are rare predisposition cancer syndrome in which CDKN2A mutations remain the most common gene association.
Materials and Methods
Clinical Data
Our patient has been referred to genetic counseling by physicians on the basis of her own clinical history described and listed in Table 1.
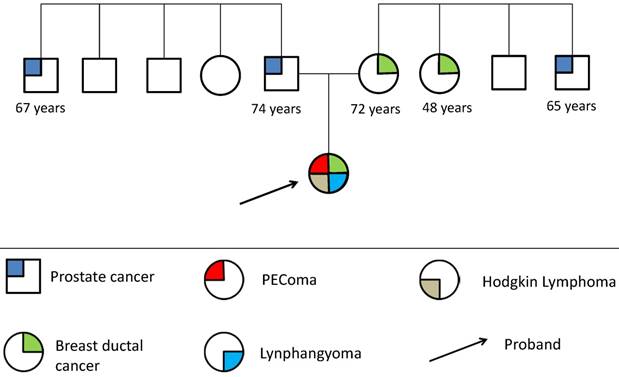
The patient is a female of 47 years old at the time of genetic visit, with a remarkable history of cancer disease: at age of 20 she was diagnosed with Hodgkin's lymphoma, treated with chemotherapy and radio-therapy, at the age of 38 she underwent to first surgical treatment for retroperitoneal lymphangioma, which is followed with a second surgical treatment for the same lesion recurrence one year later.
After three years patient was diagnosed with a retroperitoneal mass referred to malignant PEComa, finally at the age of 46 years old she was surgically treated for bilateral breast ductal in situ carcinoma (see Table 1).
Clinical history of the proband
| Patient's Age | Tumour | Treatment |
|---|---|---|
| 20 years | Hodgkin Lymphoma | Chemiotherapy and radiotherapy |
| 38 years | Retroperitoneal lymphangioma | Surgical excision |
| 41 years | Recurrence of retroperitoneal lymphangioma | Surgical excision |
| 46 years | Bilateral breast carcinoma in situ | Bilateral mastectomia |
| 47 years | Malignant PEComa. | surgical |
| 48 years | Recurrence of PEComa in the contest of Ilio-psoas muscle | surgical |
| 49 years | Patient death |
Histological analysis of retroperitoneal mass revealed classical PEComas features, positive for HMB45 antibody and also for Melan-A, microophtalmia transcription factor (Mitf) and actin.
Patient's Family history does not show occurrence of any type of rare tumor at early age. Only two cases of breast cancer disease and two cases of prostate cancer disease were reported in first degree relatives: at the age of 72 and 48 years for the breast cancer and 65 and 74 for prostate cancer respectively, overlapping epidemiological incidence data, as showed on family pedigree.
Family pedigree (fig.1) is drawn in genetic counseling, not informative for cancer risk assessment and thus for selecting specific cancer gene test among known available ones. However some patient's characteristics were suggestive of hereditary disease and it has been considered an indication for genetic testing.
Recognition of the inherited nature of the tumors is important for genetic counseling of these patients and their families.
So we analyze proband's sample DNA from blood, using a panel for multiple sequencing of 94 genes whose mutations are recognized to be engaged in the germline DNA for predisposition to tumor development.
Genetics Analysis
After the patient had signed an informed consent, a sample of peripheral blood was collected. The DNA was extracted from leukocytes using the kit EZ1 DNA Blood 200 μl, and successively analyzed by Next-Generation Sequencing (NGS) using an enrichment protocol (Illumina Trusight Cancer) for the simultaneous sequencing of 94 genes involved in the main hereditary cancer syndromes.
Familial pedigree
The panel covers the coding regions of 94 genes: AIP, ALK, APC, ATM, BAP1, BLM, BMPR1A, BRCA1, BRCA2, BRIP1, BUB1B, CDC73, CDH1, CDK4, CDKN1C, CDKN2A, CEBPA, CEP57, CHEK2, CYLD, DDB2, DICER1, DIS3L2, EGFR, EPCAM, ERCC2, ERCC3, ERCC4, ERCC5, EXT1, EXT2, EZH2, FANCA, FANCB, FANCC, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCL, FANCM, FH, FLCN, GATA2, GPC3, HNF1A, HRAS, KIT, MAX, MEN1, MET, MLH1, MSH2, MSH6, MUTYH, NBN, NF1, NF2, NSD1, PALB2, PHOX2B, PMS1, PMS2, PRF1, PRKAR1A, PTCH1, PTEN, RAD51C, RAD51D, RB1, RECQL4, RET, RHBDF2, RUNX1, SBDS, SDHAF2, SDHB, SDHC, SDHD, SLX4, SMAD4, SMARCB1, STK11, SUFU, TMEM127, TP53, TSC1, TSC2, VHL, WRN, WT1, XPA, XPC.
The DNA library was prepared starting from 50 ng of genomic DNA and was sequenced on the MiSeq sequencer (Illumina) with MiSeq Reagent Kit v2 (2x150 cycles), according to manufacturer's instructions. The sequencing reads were aligned with BWA MEM [9] against the human reference genome hg19. The resulting BAM files were processed with GATK version 3.2.2 [10] to recalibrate base qualities and realign aligned reads around indels and with MarkDuplicates to remove duplicate read-pairs, that arise as artifacts during polymerase chain reaction amplification or sequencing. To identify all variants in the samples we used Genome Analysis Toolkit Unified Genotyper (GATK, version 3.5) to search SNVs and InDel with minimum fraction of indel of 10% and minimum base quality score of 15. The genomic and functional annotation of detected variants was made by Annovar [11]. Variants were viewed manually on integrative genomics viewer (IGV) to eliminate strand bias and reduce false positive calls. Sequencing quality statistic were obtained using GATK's Depth Of Coverage tool. A second NGS analysis, following the same protocol, was performed to confirm the presence of the mutations identified.
Results
Molecular analysis by NGS reveals the presence of two deleterious germline mutations in two different genes responsible for different predisposition syndromes: Lynch syndrome (OMIM #120435) and Familial atypical multiple mole melanoma (FAMMM#155601).
For Lynch syndrome we have found a novel frameshift mutation in the PMS1 gene, c.1139dupA p.Y380_S381delinsX, that consists in a duplication of a nucleotide (A1140) in the exon9 (NM_000534) and determines an early termination of the PMS1 protein synthesis at codon 380. Only three mutations have been reported in the literature in this gene [5-6] and, although its classification as MMR gene has been recognized for a long time, its mutations were associated with Lynch syndrome only in very few cases.
In CDKN2A gene, NGS data detected the presence of c.58delG p.V20X, that consists in a deletion of a nucleotide (G58) in the exon 1β (NM_058195) and determines an early termination of the p14(ARF) protein synthesis at codon 20.
These mutations are not present in the ExAC and gnomAD database, also COSMIC database does not report these mutations [12].
CDKN2A gene encodes two different tumor suppressor proteins by alternative splicing, p16(INK4), a cyclin-dependent kinase inhibitor, and p14(ARF), an inhibitor of the p53-destablization protein MDM2[13] is an E3 ubiquitin ligase and mediates p53 degradation.
Only few mutations of CDKN2A affecting p14(ARF) but not p16(INK4) have been described in literature [14-15].
Unfortunately segregation of these mutations in the father, in the mother and in the proband's child is not available because they did not give their consent to the genetic test and to obtain the surgical specimens as well after patient's death, for this we could not verify whether mutations were present at somatic level within the tumors samples of the patient.
The other 131 exonic variants identified in the patient were filtered on the basis of the frequency in the population lower than 0.01 or unknown (Esp6500, 1000genomes and Exac03) in order to exclude them as polymorphisms. The remaining 4 variants were 2 missense mutations in TP53 (c.587G>A p.R196Q) and WRN (c.3101A>T p.Y1034F) genes and 2 synonymous mutations in EGFR (c.1509C>T p.G503G) and DICER1 (c.G1935A p.P645P) genes. All these variants have been previously described and have been classified as benign or with uncertain significance due to the lack of information.
In conclusion the coexistence of two germline mutations in two different genes, already associated to cancer predisposition syndrome, such as LS and FAMMM have been described.
Discussion
About 5% of all cancer cases are due to high penetrance, dominant germline mutations. A much larger proportion is caused by a combination of weaker susceptibility genes and/or environmental and lifestyle factors. A number of high penetrance susceptibility genes have been identified, and recently, the clinical significance of genetic germline alterations in tumors has started to be appreciated. Scientific and technological advances in genomics are revolutionizing the approach to genetic cancer risk assessment, in fact NGS technologies surely made easier and less expensive the analysis of many cancer susceptibility genes simultaneously. It has to be noted that using multi gene panels could allow the discovery of some disease-causing mutations in different genes stimulating us to reassess the nosography, the clinical and the diagnostic approach to this type of genetic syndromes in patients with evident history of multiple cancer. This will allow us to redefine the cancer risk assessment for every know cancer predisposition syndrome basing the calculation risk for every not only on a single gene mutation but on multiple gene mutations eventually identified. It is likely that further genes associated with different phenotypes in monoallelic and biallelic mutation carriers will be recognized for cancer syndromes.
Routine sequencing patients diagnosed with cancer with multiple genes panels, drive us towards a revision of the clinical classification of the tumor syndromes, better elucidating the phenotypic variability on the basis of the presence of single and/or multiple gene mutations.
Our reported case is an example about the coexistence of different rare novel mutations in different cancer predisposition genes.
The clinical data show that our patient has many tumor manifestations with aggressive characteristics, that's why we it like severe cancer predisposition syndrome.
PMS1 is not considered a classical gene causative of Lynch Syndromes, and it is not evaluated in the common clinical practice, for this reason its implication in the susceptibility to tumor development could be underestimated.
Indeed, this report shows that the deployment of NGS in medical laboratories according genetic counseling, significantly increases the throughput, providing a more accurate molecular diagnosis.
Moreover introducing the molecular analysis of multiple genes in common clinical practice help us to evaluate both clinical and molecular variability of cancer predisposition genes and thus the frequency of this conditions in the general population and more specifically in the various ethnic groups for every know cancer predisposition genes.
Finally we reported for the first time the coexistence of two common genetic syndromes in the same patient: Hereditary Nonpolyposis Colorectal Cancer (HNPCC-LS) and Familial Atypical Multiple Mole Melanoma (FAMMM #155601) (Table 2).
Known pathogenic mutations in PMS1
| Gene | Mutation | Clinical Phenotype | n° cases | Literature |
|---|---|---|---|---|
| PMS1 | p.Met394Thr ATG>ACG | 4 CRC | 1 | Qing Wang, et al., 1998 |
| P.Gly501Arg GGA>AGA | Leukemia | |||
| p.Gln233X | HNPCC | 1 | Tao Liu, et al., 2001 | |
| c.1139dupA | see Table 1 | 1 | Cassone et al. 2017 |
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wang Q. Cancer predisposition genes: molecular mechanisms and clinical impact on personalized cancer care: examples of Lynch and HBOC syndromes. Acta Pharmacol Sin. 2016Feb;37(2):143-9
2. By Aldred Scott Warthin. 1913. Classics in oncology. Heredity with reference to carcinoma as shown by the study of the cases examined in the pathological laboratory of the University of Michigan, 1895-1913. CA Cancer J Clin. 1985;35(6):348-59
3. Garber JE, Offit K. Hereditary cancer predisposition syndromes. J Clin Oncol. 2005;23:276-92
4. Lynch HT, Krush AJ. Carcinoma of the breast and ovary in three families. Surg Gynecol Obstet. 1971Oct;133(4):644-8
5. Wang Q, Lasset C, Desseigne F, Saurin JC, Maugard C, Navarro C, Ruano E, Descos L, Trillet-Lenoir V, Bosset JF, Puisieux A. Prevalence of germline mutations of hMLH1, hMSH2, hPMS1, hPMS2, and hMSH6 genes in 75 French kindreds with non polyposis colorectal cancer 1998. Hum Genet. 1999;105(1-2):79-85
6. Liu T, Yan H, Kuismanen S, Percesepe A, Bisgaard ML, Pedroni M, Benatti P, Kinzler KW, Vogelstein B, Ponz de Leon M, Peltomäki P, Lindblom A. The role of hPMS1 and hPMS2 in predisposing to colorectal cancer. Cancer Res. 2001Nov1;61(21):7798-802
7. Aronson M, Gallinger S, Cohen Z, Cohen S, Dvir R, Elhasid R, Baris HN, Kariv R, Druker H, Chan H, Ling SC, Kortan P, Holter S, Semotiuk K, Malkin D, Farah R, Sayad A, Heald B, Kalady MF, Penney LS, Rideout AL, Rashid M, Hasadsri L, Pichurin P, Riegert-Johnson D, Campbell B, Bakry D, Al-Rimawi H, Alharbi QK, Alharbi M, Shamvil A, Tabori U, Durno C. Gastrointestinal Findings in the Largest Series of Patients With Hereditary Biallelic Mismatch Repair Deficiency Syndrome: Report from the International Consortium. Am J Gastroenterol. 2016Feb;111(2):275-84
8. Hamilton SR, Liu B, Parsons RE, Papadopoulos N, Jen J, Powell SM, Krush AJ, Berk T, Cohen Z, Tetu B. et al. The molecular basis of Turcot's syndrome. N Engl J Med. 1995Mar30;332(13):839-47
9. Li C, Courtot M, Le Novère N, Laibe C. BioModels.net Web Services, a free and integrated toolkit for computational modelling software. Brief Bioinform. 2010May;11(3):270-7
10. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010Sep;20(9):1297-303
11. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010Sep;38(16):e164
12. http://exac.broadinstitute.org
13. Robertson KD, Jones PA. Tissue-specific alternative splicing in the human INK4a/ARF cell cycle regulatory locus. Oncogene. 1999Jul1;18(26):3810-20
14. Randerson-Moor JA1, Harland M, Williams S, Cuthbert-Heavens D, Sheridan E, Aveyard J, Sibley K, Whitaker L, Knowles M, Bishop JN, Bishop DT. A germline deletion of p14(ARF) but not CDKN2A in a melanoma-neural system tumour syndrome family. Hum Mol Genet. 2001Jan1;10(1):55-62
15. Binni F, Antigoni I, De Simone P, Majore S, Silipo V, Crisi A, Amantea A, Pacchiarini D, Castori M, De Bernardo C, Catricalà C, Grammatico P. Novel and recurrent p14 mutations in Italian familial melanoma. Clin Genet. 2010Jun;77(6):581-6
Author contact
![]() Corresponding author: m.cassonenl; marcocassoneit; Phone +393772867733
Corresponding author: m.cassonenl; marcocassoneit; Phone +393772867733