Oncomedicine 2018; 3:59-66. doi:10.7150/oncm.25851 This volume Cite
Research Paper
DNA Repair Response Modulation Potentiates Low Dose Cisplatin Effects in HeLa Cells
Aayushi Jain, Devashree Jahagirdar, Nilesh Kumar Sharma ![]()
Cancer and Translational Research Lab, Dr. D.Y. Patil Biotechnology & Bioinformatics Institute, Dr. D.Y. Patil Vidyapeeth, Pune, Maharashtra, India, 411033.
Received 2018-3-3; Accepted 2018-5-7; Published 2018-6-22
Abstract

Background: Cisplatin is considered as the crucial regimen of widely prescribed chemotherapy treatment to many cancer types. However, incessant increasing dose of cisplatin against many cancers including cervical cancer is known for drug resistance and side effects like nephrotoxicity and ototoxicity.
Methods: To address above problems, this paper reports using ATM kinase inhibitor (KU55933) and siRNA DNA ligase III to alter the DNA repair response in vitro low dose cisplatin treated HeLa cells. Here, authors performed cell proliferation, MTT cell cytotoxicity, annexin V/PI staining flow cytometery and PI stained cell cycle assays to substantiate observations.
Results: This paper reports that low dose cisplatin (10 µM) can show better effects in vitro growing HeLa cells in combination with KU55933. However, we included siRNA DNA ligase III along with KU55933, but data suggest that the level of DNA ligase III protein are not changed due to treatment with siRNA DNA ligase III as shown by immunocytochemistry assay. Even though, level of DNA ligase III is not altered in the present finding, use of KU55933 as combinatorial option with low dose cisplatin produce significant anti-proliferation effects (up to 35%) and also loss of cell viability including apoptotic cell death (up to 30%) in treated HeLa cells.
Conclusion: This study suggests that KU55933 in combination with low dose cisplatin may be a good option to bring anti-proliferation and loss of viability in HeLa cells and may be explored in other cancer cell lines as well.
Introduction
At global level, cervical cancer is considered as the second most happening cancer types [1]. Chemotherapy is one of the therapies of cancer where the chemical compounds are used as drug to target the genes or proteins [2-8]. There are several compounds used as anticancer drugs such as alkylating agents including platinum based drugs [9-13]. In spite of certain success, these treatment options face limitations and huddles such as drug resistance, side effects as nephrotoxicity heart failures and relapse of cancer [6, 9-13]. The mechanism of action of cisplatin revolves around binding at the N7 guanine residue in dsDNA, thereby forming 1, 2 intra-strands [2, 14-17]. Adducts formed by Cisplatin are recognized by the repair pathways like NER (Nucleotide Excision Repair), BER (Base Excision Repair) and MMR (Mismatch Repair) [2,10,14,15]. These repair pathways involve different recognition proteins to bind to the damaged DNA, after which they initiate either repair or apoptosis [2,18,19].
Among many specific DNA repair proteins, ATM kinase and DNA ligase III are suggested to contribute in cisplatin mediated DNA repair system that leads to the better cell survival instead of cell death [4,8,9,10,12,19,20,21]. This article attempts to establish the importance of inhibiting ATM kinase and also DNA ligase III for enhanced effects of low dose cisplatin in HeLa cells. Hence, present paper emphasizes the scope of using low dose cisplatin to show fewer side effects and at the same time with better efficacy in combinatorial options with DNA repair protein inhibitors.
Material and Methodology
Materials
Cell culture reagents were purchased from Invitrogen India Pvt. Ltd. and Himedia India Pvt. Ltd. HeLa cells were procured from National Centre of Cell Science (NCCS), Pune, India. Cisplatin and KU55933 were procured from Cayman Chemical chemical, USA. Propidium iodide and apoptotic assay kit was purchased from ThermoFisher Scientific, USA. All other chemicals were of analytical grade and obtained from Himedia India Pvt. Ltd and Merck India Pvt. Ltd.
Cell Line Maintenance and Seeding
HeLa cells were cultured and maintained in DMEM (Dulbecco's Modified Eagles Medium) (Himedia) with high glucose supplemented with 10% heat inactivated NBCS (New Born Calf Serum) (Himedia)/penicillin (100 units/ml)/streptomycin (100 µg/ml) at 37°C in a humidified 5% CO2 incubator.
Cell Proliferation and Viability Assay
Cytotoxicity assays were carried out in 6 well plates. 1.5 x 105 cells were seeded and after 16 h, respective treatment of drug and inhibitor was subjected to cells and incubated in CO2 incubator at 37ºC for 72 h. The following treatment conditions were used for HeLa cells proliferation assay as DMSO control, cisplatin (10 µM), siRNA DNA ligase III (20 picomols of siRNA duplex) [sc-72079, Santacruz Biotechnology] + KU55933 (2.5 µM) and cisplatin (10 µM) + siRNA DNA ligase III (20 picomols of siRNA duplex) + KU55933 (2.5 µM). In this paper, it is worth to mention that use of siRNA DNA ligase III was preceded without the transfection reagents due to discernible toxicity from the transfection reagents alone. In this case authors considered the possibility of entry of exogenous siRNA after making complexes with extracellular vesicles present in the serum components of used culture medium. After completion of 72 h, HeLa cells were harvested and total cell count and cell viability estimation were carried out by trypan blue dye exclusion assay and hemocytometer counting.
Mtt Cell Cytotoxicity Assay
HeLa cells were seeded into 96 well flat bottom plate with the seeding density of 10000 cells per well in a final volume of 200 µl. Next, HeLa cells were allowed to adhere for next 16-18 h. After 16 h, cells were treated in triplicates for treatment combinations as mentioned in the cell proliferation assay. In this experiment, HeLa cells were incubated with above described treatment conditions for 72 h at 37°C. At the end of experiment, medium from wells was aspirated. Further, HeLa cells were treated with 100 µl of RPMI medium to avoid interference during absorbance. Then, 10 µl of (Thiazolyl blue tetrazolium) MTT solution was added to each respective well. Next, cells were then incubated for 2-4 h in dark. At the end of incubation and observation of purple color, 100 µl of isopropanol was added to each well. The plate was gently stirred and pipetted in and out to allow the crystals to be dissolved. The cells were incubated for 2 h in dark and the absorbance was measured by using ELISA reader (ThermoFisher Scientific) at 570 nm and at a reference wavelength of 650 nm.
Fluorescent Microscopy
Prior to seeding HeLa cells onto a 6 well plate the wells were coated with 0.4% gelatin immersed with a sterile coverslip. This was incubated at 4°C and later cells were seeded following the drug treatment options as mentioned in case of cell proliferation assay. At the end of treatment, HeLa cells were washed with PBS. Fixation of HeLa cells was carried out with 4% formaldehyde followed by cell permeabilizing with Triton X. Blocking of the non-specific binding sites was carried out by 4% blocking buffer and rabbit primary antibody anti-DNA Ligase III was added (1:10000) and followed by secondary anti-rabbit Alexa flour 568. Then after, HeLa cells were also stained with DAPI. After necessary incubation, HeLa cells were observed under fluorescent microscope (Zeiss) at 40X magnification using DAPI and Alexa flour 568 red filter.
Flow Cytometry Based Cell Viability and Apoptosis Determination Assay
HeLa Cells were subjected to desired treatment conditions as mentioned in above cell proliferation assay. HeLa cells were harvested at the end of 72 h of treatment and cells were washed with PBS. The pelleted HeLa cells were suspended in 1X Annexin Binding Buffer (ABB) and FITC Annexin V was added followed by PI (100 µg / ml) and incubated for 15 min at the room temperature. Then, cells were analyzed on BD FACSJazz Cytometer to count the live and dead cells. At least, 10,000 events were collected and analyzed per measurement.
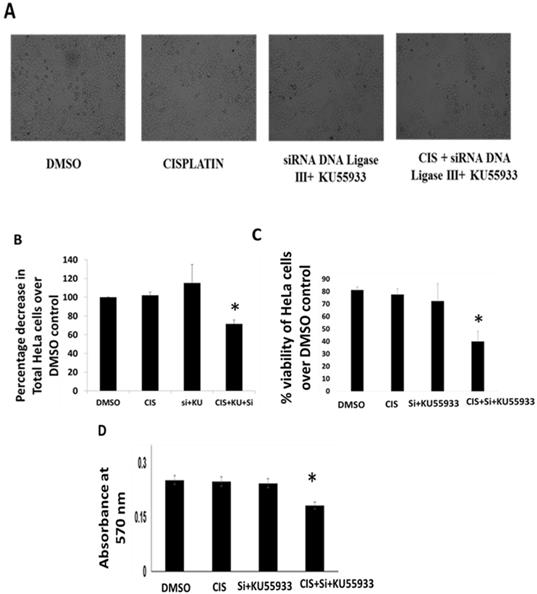
Cell viability and growth inhibition assay on HeLa cells. (A). Photomicrography images of HeLa cells were captured by inverted microscope using 10X objective in order of DMOS control, cisplatin (CIS) (10 µM), siRNA DNA ligase III (si), (20 picomols of siRNA duplex) + KU55933 (2.5 µM) and cisplatin (CIS) (10 µM) + siRNA DNA ligase III (si), (20 picomols of siRNA duplex) + KU55933 (2.5 µM). (B) Graphical representation of the decrease in total HeLa cell count over DMSO control. (C) Graph represents the percentage HeLa cell viability over DMSO control. (D) This bar graph represents the absorbance value using MTT assay. The data are represented as mean ± SD. Each experiment was conducted independently three times. The bar graph without any asterisk denotes that there is no any significant difference compared to DMSO control. * Significantly different from DMSO control at P-value < 0.05.
Cell Cycle Analysis
HeLa cells were plated and treated with the desired drug and inhibitor treatment options as described in previous cell proliferation assay section. At the end of treatment for 72 h, HeLa cells were harvested and washed with ice-cold 1X PBS for 3 times. After washing the cells were subjected to fixation by 70% ethanol with incubation at 4°C for 30 min. The fixated cells were passed through cell strainer to eliminate the clumped cells. Further, the cells were washed with PBS and they were subjected to RNase (5µg/ml) and PI (10µg/ml). Finally, stained cells were analysed on BD FACSJazz using blue laser.
Statistical Analysis
Data shown are presented as the mean ± SD of at least three independent experiments; differences are considered statistically significant at P < 0.05, using a Student's t-test.
Results
Anti-proliferative and loss of viability
To evaluate the potentiation of low dose cisplatin effects in vitro HeLa cells, authors report the combined effects of KU55933 and siRNA DNA ligase III for anti-proliferative and bring cell cytotoxicity in HeLa cells. By using Trypan blue dye exclusion and MTT assay, authors see minimal anti-proliferative and toxicity due low dose cisplatin (10 µM). Interestingly, combined use of KU55933 and siRNA DNA ligase III along with low dose cisplatin showed significant level of reduction in the HeLa cell count and also loss in viability up to 30% (Figure 1A-D). Data from Trypan blue dye exclusion and MTT assay are showing coherence to each other and suggest less proliferation and loss of viability of HeLa cells. However, authors would like to clarify that due to technical incompatibility of siRNA DNA ligase transfection protocol, there is not significant reduction in the DNA ligase III protein level. Therefore, data suggest that only ATM kinase inhibition by KU55933 (2.5 µM) might have worked in the present treatment condition and appears to be potentiating the effects of low dose cisplatin with reduction in proliferation capacity and enhanced toxicity in HeLa cells. However, from this experiment, it is not clear about the observed effects in HeLa cells either through cell cycle arrest or apoptotic cell death.
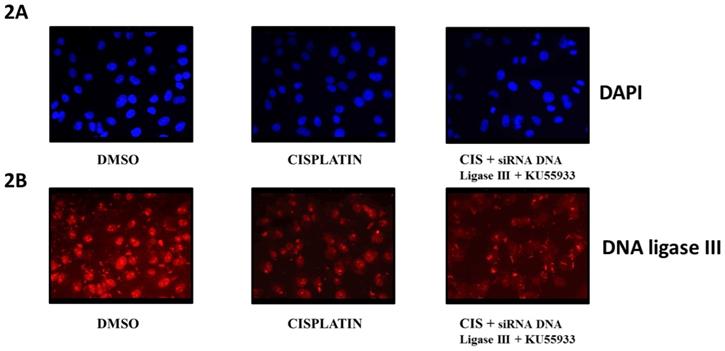
Immunofluorescent microscopy of DNA ligase III protein level in HeLa cells. This diagram represents the fluorescent microscopy images of HeLa cells stained with DAPI [2A] and anti-DNA Ligase III antibody and probed with secondary antibody AlexaFluor 568 [2B]. Here, HeLa cells were treated as DMOS control, cisplatin (CIS) (10 µM), siRNA DNA ligase III (si), (20 picomols of siRNA duplex) + KU55933 (2.5 µM) and cisplatin (CIS) (10 µM) + siRNA DNA ligase III (si), (20 picomols of siRNA duplex) + KU55933 (2.5 µM). These photographs were captured at 40X using Zeiss Fluorescent microscope. Each experiment was conducted independently three times.
Immunocytochemistry assay
To evaluate the extent of effects of siRNA DNA ligase III in HeLa cells, we submitted treated HeLa cells for immunocytochemistry by using anti-DNA ligase III primary antibody and observed at 40X using fluorescent microscope (Figure 2B). In another panel, DAPI stained HeLa cells images are given (Figure 2A). Due to issue in siRNA transfection protocol, we do not find significant reduction in the DNA ligase III protein expression in the HeLa cells treated with siRNA DNA ligase III in combination with KU55933 and cisplatin. Analysis on stained HeLa cells treated with combinations of cisplatin and KU55933 confirms the reduction of HeLa cells as observed in case of Trypan blue dye exclusion and MTT assays. At the same time, present data is not able to suggest the combinatorial use of siRNA DNA ligase III due to absence of significant reduction of DNA ligase III protein levels in the treated HeLa cells.
Cell viability and apoptosis assay by flow cytometery
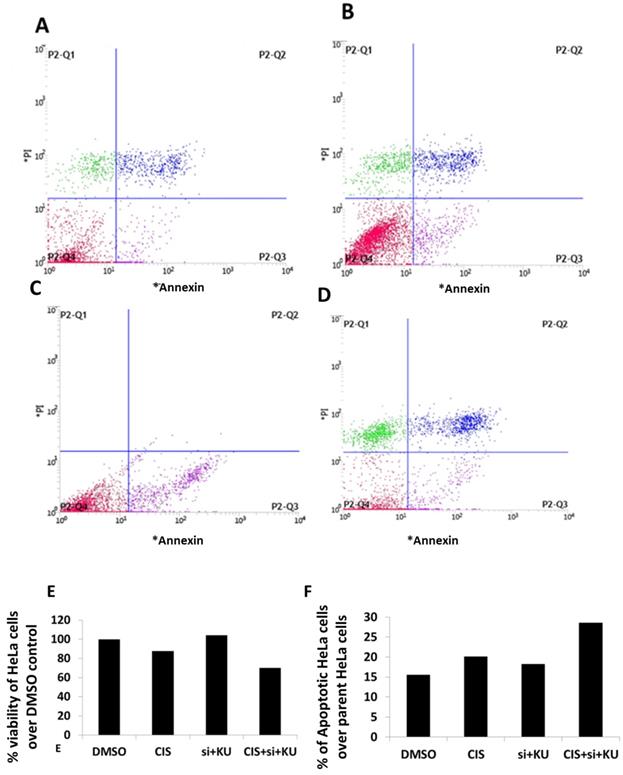
To estimate the extent of apoptosis and loss of viability in HeLa cells treated concomitantly with low dose cisplatin, siRNA DNA ligase III and KU55933, the use of annexin V/FITC and PI staining method is preferred. The flow cytometer graphs are presented showing the intensity of HeLa cells stained with PI and annexin V/FITC (Figure 3-D). The analysis of flow cytometer graph is shown as percentage of viability of HeLa cells treated with DMSO and low dose cisplatin, siRNA DNA ligase III and KU55933 (Figure 3E). We also estimated the extent of percentage of apoptotic cells in the parent populations of HeLa cells treated with DMSO and low dose cisplatin, siRNA DNA ligase III and KU55933 (Figure 3F). Data show to some extent up to 30% loss of viability of HeLa cells treated with combination of cisplatin, siRNA DNA ligase III and KU55933 over the DMSO and drugs alone. On the other side, it also appears that slight increase in the apoptotic cells in Hela cells treated with concomitant use of low dose cisplatin, siRNA DNA ligase III and KU55933 in comparison to DMSO and drugs alone.
Cell cycle analysis
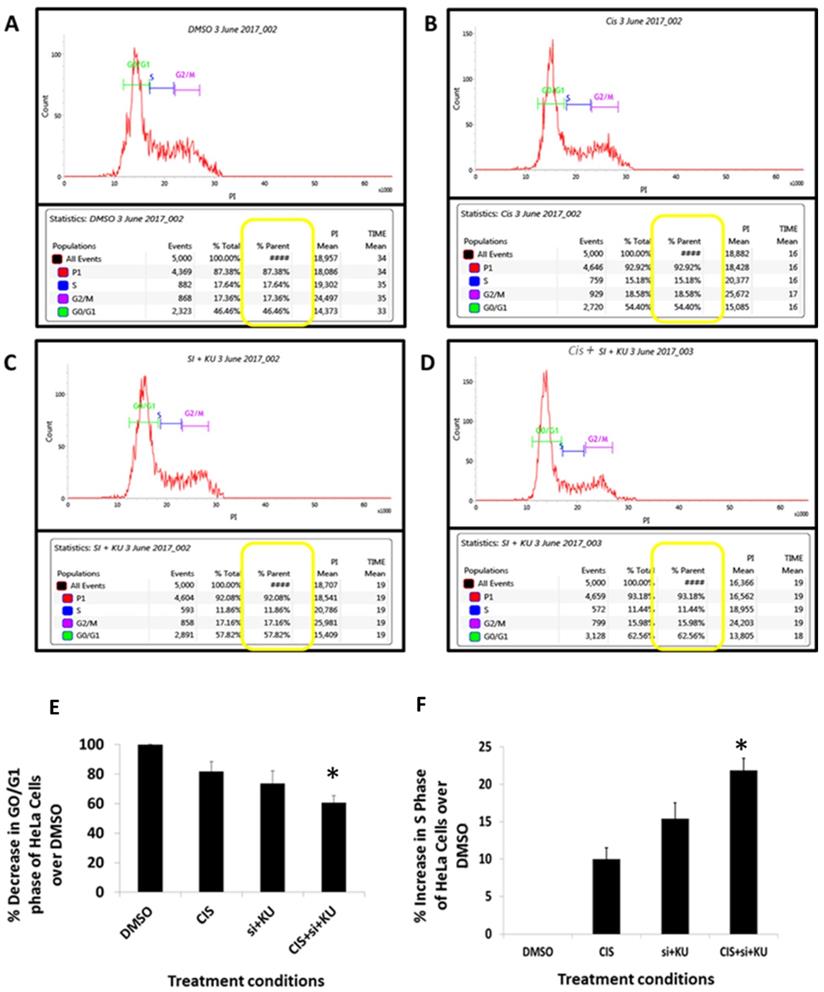
To evaluate the potential of combined use of cisplatin, siRNA DNA ligase III and KU55933 against HeLa cells, the pattern of cell cycle stages in HeLa cells were studied using PI staining flow cytometer assay (Figure 4A-D). The PI intensity calculated by the flow cytometer is proportional to the distribution of DNA in phases like G0/G1, S and G2/M. Combinatorial treatment of Cisplatin, KU55933 and siRNA DNA ligase III depicts that proportion of G0/G1 has significantly decreased compared to the DMSO control almost by 30% (Figure 4E). At the same time, percentage of S phase of HeLa cells treated with cisplatin, siRNA DNA ligase III and KU55933 were appreciably increased by 22% compared to the DMSO control (Figure 4D).
Apoptotic cell death estimation in HeLa cells using annexin V/PI staining method. (A) Data represents HeLa cells stained with PI and annexin-V conjugated with FITC and treated with DMSO control. (B) Data represents HeLa cells stained with PI and annexin-V conjugated with FITC and treated with (CIS) (10 µM). (C) Data represents HeLa cells stained with PI and annexin-V conjugated with FITC and treated with siRNA DNA ligase III (si), (20 picomols of siRNA duplex) + KU55933 (2.5 µM). (D) Data represents HeLa cells stained with PI and annexin-V conjugated with FITC and treated with cisplatin (CIS) (10 µM) + siRNA DNA ligase III (si), (20 picomols of siRNA duplex) + KU55933 (2.5 µM). (D). this bar graph represents the percentage decrease in HeLa cell viability over DMSO control. (D). This bar graph shows the total apoptotic HeLa cells over DMSO control. Data presented in this figure represents the mean of three replicates and processed for single time flow cytometer analysis.
Discussion
Cancer being one of the most devastating diseases is widely spreading all around the globe. There are several anti-cancer drugs and also synergistic drug regimens that target intracellular pathways including DNA damage response pathways [2,3,4,7,8,20]. This combination of drugs and inhibitors produces plethora of responses including cell cycle arrest, apoptosis and other cell death process [2-4,7-8,20].
Flow cytometer PI staining cell cycle study. HeLa cells were incubated for 72 h. A: DMOS control. B: cisplatin (CIS) (10 µM), C: siRNA DNA ligase III (si), (20 picomols of siRNA duplex)+ KU55933 (2.5 µM). D: cisplatin (CIS) (10 µM) + siRNA DNA ligase III (si), (20 picomols of siRNA duplex) + KU55933 (2.5 µM). E: Percentage of G0/G1 phase of cell cycle over DMSO control. F: Percentage decrease in S phase of cell cycle over DMSO control. Cells were then harvested by trypsinization, fixed, and stained with propidium iodide for cell cycle analysis by flow cytometry. The data are represented as mean ± SD. Each experiment was conducted independently three times. The bar graph without any asterisk denotes that there is no any significant difference compared to DMSO control. * Significantly different from DMSO control at P-value < 0.05.
The well-known platinum based drug, cisplatin, is being used from decades for carcinoma treatment including cervical cancer. The major issue concerned with cisplatin as a chemotherapeutic option with excessive over dose leading to drug resistance and patients secondary pathophysiological conditions including secondary tumor [2,5,6,10,14,16,22]. The reasons behind cisplatin resistance and efficacy issues may be due to decreased accumulation of drug inside the cell, altered DNA repair or increased efflux of drug [2,10,14,16,23,24]. One such outlook is use of the small molecule inhibitors targeted to the DNA repair response proteins along with the drug cisplatin [4,7,8,20,24]. These small molecule inhibitors such as KU-55933, SCR7, L189 and siRNA DNA Ligase III are reported among with cisplatin thereby potentiating the responsiveness of cisplatin drug [4,7,20-26].
There are reports on the efficacious action of concomitant treatment with genotoxic drugs and DNA repair inhibitors in many cancer types [6,9-13]. At the same time, these combinatorial drug options show varied molecular and cellular responses including anti-proliferative, apoptosis and autophagy and loss of viability [6,9-13].
Therefore, present paper demonstrates the clear and significant reduction in total HeLa cell count and also decreased viability. Trypan blue dye exclusion and MTT assays are basic and simple cell based assay to study anti-proliferative and cell viability in vitro growing cells. The use of this assay is also appreciated to determine the total cell content and in turn to assess the cell proliferation rate [27-29]. The degree of observed anti-proliferative and loss of viability is almost comparable to other reported studies using cisplatin and other classes of synergistic drugs and inhibitors [11,13]. Interestingly, total cell proliferation and MTT based cell viability data is well supported from the annexin V/PI staining apoptotic cell death assay. In case of annexin V/PI staining results, we found loss of viability up to 30%, that is in consonance with the Trypan blue dye exclusion and MTT based assay. At the same time, data point out the presence of apoptosis to some extent, but the possibilities of other cell death mechanisms such as autophagy may not be ruled out and there is a need to further investigate in future. Further, cell cycle analysis is also in coherence with the other cell based assays and finding shows that HeLa cells treated with combinations of cisplatin and KU55933 are arrested in G0/G1 phase of HeLa cell cycle and also appreciable reduction in S phase of cell cycle. However, data do not suggest in the alteration of G2/M phase cell cycle due these treatment conditions.
In past years, there are appreciable evidences on the use of other form of combinatorial drug regiments to enhance the toxicity of low dose cisplatin and at the same time removing issues related to high dose effects in cancer treatment. In a low dose cisplatin based toxicity study in HeLa cells, authors show that Interferon β combined with cisplatin can be useful for better effectiveness [11]. Another evidence indicates that low dose of cisplatin in HeLa cells can be forced to go for autophagy by the combinatorial use of rapamycin drug [13].
Reports also mention that action of DNA ligase III and ATM kinase are interrelated in a way where the ATM is responsible for the recruiting the ligase which is been further involved in cdk2 cell cycle proteins and hence participate co-operatively in repairing the DNA [30,31]. However, in our case, we do not find any clear role of DNA ligase III in the combinatorial treatment to HeLa cells, as reduction in DNA ligase III protein level is not achieved up to the desired level. Therefore, authors exclude the potential role of DNA ligase III and suggest that KU55933 can show significant anti-proliferative and to some extent bring apoptotic cell death to support the loss of HeLa cell viability. Authors do not exclude the contribution of autophagy mediated cell death for the clearly observed loss of HeLa cell viability due to combined effects of cisplatin and KU55933. The role of ATM kinase in NER mediated DNA repair responses to cisplatin action in cancer cell are reported for better survival of cancer cell including HeLa cell. Hence, the present data support the existing view that by targeting ATM kinase using small molecule inhibitor may put HeLa cell under GO/G1 cell cycle arrest. Additionally, authors find the additional observations in the form of blockade of HeLa cells to enter into S phase of cell cycle. This observation needs to be investigated in future.
Conclusion
In conclusion, present research work supports the combinatorial drug approaches including cisplatin at low dose and KU55933 an ATM kinase inhibitor to bring appreciable cell blockade of HeLa cell proliferation and loss of viability. In the current understanding, these drugs/inhibitors approaches sound logical as the role of inter and intra-tumor heterogeneity is highly known to impact the drugs/inhibitors responses. Therefore, in future use of cisplatin and KU55933 should be standardized and tested based on the personalized medicine where patients are having high ATM kinase expression.
Acknowledgements
The authors acknowledge financial support from DST-SERB, Government of India, New Delhi, India (SERB/LS-1028/2013) and Dr. D. Y Patil, Vidyapeeth, Pune, India (DPU/05/01/2016). The authors acknowledge Central Research Facility, DPU, Pune for flow cytometer and fluorescent microscopy experimentation facilities.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Akbari M, Keijzers G, Maynard S. et al. Overexpression of DNA ligase III in mitochondria protects cells against oxidative stress and improves mitochondrial DNA base excision repair. DNA repair. 2014;16:44-53
2. Albarakati N, Abdel-Fatah TM, Doherty R, Russell R, Agarwal D, Moseley P. et al. Targeting BRCA1-BER deficient breast cancer by ATM or DNA-PKcs blockade either alone or in combination with cisplatin for personalized therapy. Mol Oncol. 2015;9(1):204-17
3. Albarakati N, Abdel-Fatah TM, Doherty R, Russell R, Agarwal D, Moseley P, Perry C, Arora A, Alsubhi N, Seedhouse C. et al. Targeting BRCA1-BER deficient cancer by ATM or DNA-PKcs blockade either alone or in combination with cisplatin for personalized therapy. Mol Oncol. 2015;9(1):204-17
4. Amable L, Fain J, Gavin E, Reed E. Gli1 contributes to cellular resistance to cisplatin through altered cellular accumulation of the drug. Oncology Reports. 2014;32(2):469-474
5. Basu A, Krishnamurthy S. Cellular Responses to Cisplatin-Induced DNA Damage. Journal of Nucleic Acids. 2010. 2013 67
6. Bhatkar D, Kumar J, Purohit S, Jahagirdar D, Sharma NK. ATM kinase inhibitor KU-55933 contribution in cisplatin mediated HeLa proliferation. International Journal of Pharmacology and Toxicology. 2016;4(2):201-207
7. Colton SL, Xu XS, Wang YA, Wang G. The involvement of ataxia-telangiectasia mutated protein activation in nucleotide excision repair-facilitated cell survival with cisplatin treatment. J Biol Chem. 2006;281(37):27117-25
8. Curtin N. J. DNA repair dysregulation from cancer driver to therapeutic target. Nature Reviews Cancer. 2012;12(12):801-817
9. Dong Z, Tomkinson AE. ATM mediates oxidative stress-induced dephosphorylation of DNA ligase IIIα. Nucleic Acids Research. 2006;34(20):5721-5279
10. Ethiraj P, Veerappan K, Samuel S, Sivapatham S. Interferon β improves the efficacy of low dose cisplatin by inhibiting NF-κB/p-Akt signaling on HeLa cells. Biomed Pharmacother. 2016;82:124-32
11. Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, Castedo M, Kroemer G. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31(15):1869-83
12. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-74
13. Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461(7267):1071-8
14. Jain A, Jahagirdar D, Nilendu P, Sharma NK. Molecular approaches to potentiate cisplatin responsiveness in carcinoma therapeutics. Expert Rev Anticancer Ther. 2017;17(9):815-825
15. Kristine S. Louis and Andre C. Siegel. Cell Viability Analysis Using Trypan Blue: Manualand Automated Methods. A book chapter in Martin J. Stoddart (ed.), Mammalian Cell Viability: Methods and Protocols, Methods in Molecular Biology. 2011. vol. 740, DOI 10.1007/978-1-61779-108-6_1, © Springer Science+Business Media, LLC. 2011
16. Kumar A, Purohit S, Sharma NK. Aberrant DNA double-strand break repair threads in breast carcinoma: Orchestrating genomic insults survival. Journal of Cancer Prevention. 2016;21(4):227-234
17. Li YT, Chua MJ, Kunnath AP, Chowdhury EH. Reversing multidrug resistance in breast cancer cells by silencing ABC transporter genes with nanoparticle-facilitated delivery of target siRNAs. International Journal of Nanomedicine. 2012;7:2473-2481
18. Liu F, Suryadi J, Bierbach U. Cellular Recognition and Repair of Monofunctional-Intercalative Platinum-DNA Adducts. Chem Res Toxicol. 2015;28(11):2170-8
19. Lyons A.B, Blake S.J. and Doherty, K.V, Flow cytometric analysis of cell division by dilution of CFSE and related dyes. Current protocols in cytometry. 2013 Chapter 9:Unit9.11. doi: 10.1002/0471142956.cy0911s64
20. Murai J. Targeting DNA repair and replication stress in the treatment of ovarian cancer. Int J Clin Oncol. 2017;22(4):619-628
21. Piotr Widlak Æ Monika Pietrowska Æ Joanna Lanuszewska. The role of chromatin proteins in DNA damage recognition and repair. Histochem Cell Biol. 2006;125:119-126
22. Salgia R, Kulkarni P. The Genetic/Non-genetic Duality of Drug 'Resistance' in Cancer. Trends Cancer. 2018;4(2):110-118
23. Schnitt SJ. Classification and prognosis of invasive breast cancer: from morphology to molecular taxonomy. Modern Pathology. 2010:23 S60-S64
24. Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22(47):7265-79
25. Simsek D, Jasin M. DNA ligase III: A spotty presence in eukaryotes, but an essential function where tested. Cell Cycle. 2011;10(21):3636-3644
26. Soza S, Leva V, Vago R. et al. DNA Ligase I Deficiency Leads to Replication-Dependent DNA Damage and Impacts Cell Morphology without Blocking Cell Cycle Progression. Molecular and Cellular Biology. 2009;29(8):2032-2041
27. Strober W. Trypan blue exclusion test of cell viability. Curr Protoc Immunol. 2001 Appendix 3:Appendix 3B. doi: 10.1002/0471142735.ima03bs21
28. Vecchio D, Frosina G. Targeting the Ataxia Telangiectasia Mutated Protein in Cancer Therapy. Curr Drug Targets. 2016;17(2):139-53
29. Wheate NJ, Walker S, Craig GE, Oun R. The status of platinum anticancer drugs in the clinic and in clinical trials. Dalton. 2010;39(35):8113-27
30. Xuerui Chen, Li Zhang, Shiping Ding, Qunfang Lei, Wenjun Fang. Cisplatin combination drugs induce autophagy in HeLa cells and interact with HSA via electrostatic binding affinity. RSC Adv. 2017;7:22270
31. Zhou S, Huang Q, Zheng S, Lin K, You J, Zhang X2. miR-27a regulates the sensitivity of breast cancer cells to cisplatin treatment via BAK-SMAC/DIABLO-XIAP axis. Tumour Biol. 2016;37(5):6837-45
Author contact
![]() Corresponding author: Dr. Nilesh Kumar Sharma, Associate Professor, Cancer and Translational Research Lab, Department of Biotechnology, Dr. D. Y. Patil Biotechnology & Bioinformatics Institute, Dr. D. Y Patil Vidyapeeth, Pune, MH, 411033. Email: nilesh.sharmaedu.in; Phone: +91-7219269540
Corresponding author: Dr. Nilesh Kumar Sharma, Associate Professor, Cancer and Translational Research Lab, Department of Biotechnology, Dr. D. Y. Patil Biotechnology & Bioinformatics Institute, Dr. D. Y Patil Vidyapeeth, Pune, MH, 411033. Email: nilesh.sharmaedu.in; Phone: +91-7219269540