Oncomedicine 2018; 3:74-81. doi:10.7150/oncm.26116 This volume Cite
Review
siRNA: A Promising New Tool for Future Breast Cancer Therapy
Amy Huynh1, Chikezie O. Madu2, Yi Lu3 ![]()
1. Department of Biology and Advanced Placement Biology, White Station High School, Memphis, Tennessee 38117. USA. amyhnh@gmail.com
2. Departments of Biology and Advanced Placement Biology, White Station High School, Memphis, Tennessee 38117. USA. maduco@scsk12.org
3. Departments of Pathology and Laboratory Medicine, University of Tennessee Health Science Center, Memphis, TN 38163, USA. ylu@uthsc.edu
Received 2018-3-16; Accepted 2018-5-7; Published 2018-9-1
Abstract

Breast cancer remains a major health concern as it continues to be both the leading cause of cancer-related deaths and most commonly diagnosed form of cancer among women. Recently, RNAi (RNA interference)-based gene therapy has gained attention for its sequence-specific mechanism of gene silencing. RNAi may be induced with the use of short interfering RNAs (siRNAs), which are generated from double-stranded RNA (dsRNA) precursors. Although siRNA has its limitations, siRNA shows enormous potential as a promising tool for the regulation of disease-associated genes, including oncogenes and tumor suppressor genes. In this review, we will discuss the approaches, implications, and challenges of siRNA-mediated RNAi gene therapy regarding various targets of breast cancer.
Keywords: siRNA, Breast Cancer Therapy
Introduction
Breast cancer continues to be a public health dilemma. Worldwide, breast cancer is cited as the most commonly diagnosed form of cancer and the leading cause of cancer-related death among women. High-income countries, including countries in North America, Australia, and Western and Northern Europe, continue to exhibit the highest breast cancer incidence rates; this disparity among other regions of the world reflects differences in the availability of treatments and the various trends in risk factors. [1] Major factors associated with an increased risk of breast cancer development include individual lifestyle, family history of breast cancer, and genetic predisposition. [2,3] Regarding individual lifestyle, several studies have yielded consistent results suggesting that obesity, smoking, and alcohol consumption have a positive correlation with an increased risk of developing breast cancer. [2] Women with a family history of breast cancer appear to have an increased risk of developing the disease; in a study, compared to women with no affected relatives, women with one, two, or three or more first degree relatives affected with breast cancer had risk ratios of, respectively, 1.80, 2.93, and 3.90, suggesting a positive correlation with breast cancer development. [4] Although approximately 20% to 25% of breast cancer patients show a family history of breast cancer, only 5% to 10% of breast cancer cases exhibit an autosomal dominant inheritance, with high-risk predisposition alleles conferring as high as an 85% lifetime risk of developing breast cancer. [3]
Recent advancements in gene therapy have highlighted small interfering RNA (siRNA) as a promising tool for application in breast cancer therapy. siRNAs are double-stranded RNAs that participate in RNA interference (RNAi), which is a posttranscriptional mode of gene regulation. [9] siRNAs act to regulate endogenous genes and to protect the genome against foreign or invasive nucleic acids in both the somatic cells and germ cells of many eukaryotic species. Specifically, siRNAs are incorporated into the RNAi-induced silencing complex (RISC); the siRNA component of RISC recognizes its target mRNA by Watson-Crick base pairing, and this complex mediates the sequence-specific binding and cleavage of mRNA. [5,6] Within mammalian cells, siRNAs are produced from the cleavage of double-stranded RNA precursors by the RNase III endonuclease Dicer, but siRNAs may also be synthesized by chemical or biochemical methods and delivered to cells [7,8]. Mouse cells carrying a null allele of Dicer have been observed assembling siRNA into functional RISCs, indicating that Dicer is neither involved in nor required for the assembly of functional RISCs in mammals. [10]
The typical pathway of siRNA. siRNA is generated from dsRNA precursors, which are cleaved by Dicer to form siRNAs. The resultant siRNAs are then incorporated into RISCs, which cleave the target mRNA and effectively silence the expression of a particular gene. (Adapted from [58])
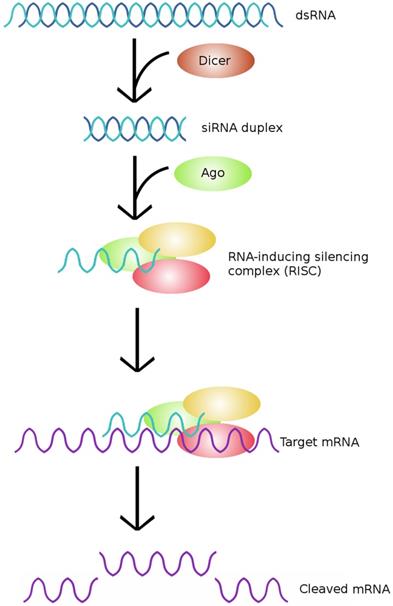
Although the application of siRNAs in breast cancer therapy is very promising, certain limitations must be considered before its use in biomedical applications. One of the major limitations to siRNA delivery is siRNA degradation; siRNAs are readily degraded by the abundant RNAses in both the intracellular and extracellular space, and naked siRNA in serum has been demonstrated to have a half-life of between a few minutes to an hour. [5] Additionally, compared to DNA-based RNAi drugs, directly using synthetic siRNA effectors may result in a potent but short-lived silencing of a gene, which calls for the use of more resources for more frequent treatments. [8]
Structure and Function of siRNA
siRNA molecules are small, double-stranded RNA (dsRNA) molecules that are roughly 19 to 30 nucleotides long. [9] Each siRNA strand possesses characteristic 5'-phosphate and 3' hydroxyl termini, as well as 2 to 3 nucleotide-long 3' overhangs. [11] In mammalian cells, longer dsRNA precursors, originating from either the environment or synthesized within the cell itself, are cleaved by the RNaseIII endonuclease Dicer to produce siRNAs, which then associate with nucleases and other proteins to form RISCs, as depicted in Figure 1. [5,6,24] Additionally, for each siRNA duplex, one of the two strands, the guide strand, is incorporated into the active RISC, while the other strand, the passenger strand, is cleaved by Argonaute2 (Ago2) and hence discarded. [12] Argonaute family proteins are an essential component of RISCs—in fact, Argonaute family proteins are ubiquitous among all RNAi-related pathways, and they are directly responsible for the slicer activity of the RISC. [13]
The siRNA portion of the RISC recognizes its target miRNA sequence by Watson-Crick base pairing; that is, the RISC targets mRNAs that possess regions complementary to the guide strand sequence of siRNA that has been incorporated into the RISC. [6,18] Following this, Argonaute proteins carry out the slicer activity of the RISC, cleaving the target mRNA, and the now-fragmented mRNA is then targeted by cellular exonucleases and finally degraded, inhibiting the translation of the target gene. [13,16] The newly generated 3' ends of the mRNA fragments also serve as a substrate for polyuridylation, which promotes exonucleolytic targeting. [17] siRNA silencing is also readily reprogrammable and functions in the adaptive immune response; when encountering new threats from novel invaders, the genome is able to take advantage of the situation by co-opting the foreign sequences into the siRNA mechanism, ceasing the expression of invasive genes. [6] Several other forms of RNA used in RNAi exist, including short hairpin RNA (shRNA) and Piwi-interacting RNA (piRNA), but siRNA is most often used for RNAi in therapeutic applications. [5]
The expression of shRNA, however, may be used to introduce siRNA into cells. shRNA can be introduced into a cell by plasmid vectors and transcribed under control of RNA polymerase-II or RNA polymerase-III promoters. The transcribed shRNA folds into a structure similar to that of a siRNA duplex, and Dicer processes the transcribed shRNAs into siRNAs. [27]
Advantages of RNAi-Based Gene Therapy
Because of its ability to carry out sequence-specific gene silencing, RNAi is a highly attractive approach to gene therapy. Theoretically, a single mRNA sequence would be sufficient enough to develop antisense drugs, and oncogenes and tumor suppressor genes, along with any other disease-associated genes, would be made vulnerable to RNAi-mediated silencing. [24] RNAi is also especially potent compared to other antisense agents, such as ribozymes and DNA oligonucleotides, meaning that lower concentrations of RNAi effector molecules may function at either the same efficiency or even more efficiently than DNA oligonucleotides or ribozymes. [14]
Challenges of Using siRNA
Although siRNA possesses great potential as a promising tool for target-specific gene silencing, certain limitations arise due to its various properties. Unmodified (naked) siRNAs are extremely unstable intravascularly; because of this, unmodified siRNAs are often susceptible to serum RNase A-type nucleases and quick renal clearance and consequently have a short half-life of approximately a few minutes to an hour. [5,20] However, chemical modifications may be used to increase the stability of siRNAs in serum. Modification of the backbone or of the sugars on siRNA using 2'-O-methyl and 2'-deoxy-2'-fluoro RNA has been shown to increase siRNA resistance to nucleases, while modification using phosphorothioate linkages has been shown to improve siRNA half-life. [5,15] Another solution is the use of lipid-based nanocarriers as a safe and efficient method of delivery to target cells, since lipid-based nanocarriers are poorly permeable but easily degradable within the target cell. [26]
Additionally, unmodified siRNAs present potential toxicities, with one being off-target effects. Genome-wide monitoring of gene expression using microarrays has demonstrated that cells treated with siRNA exhibit off-target silencing of a sizable number of genes; analysis further indicated that an 11-nucleotide match between the off-target and siRNA was sufficient enough to result in gene knockdown. [21] Later work suggests that of the experimentally verified off-targets, a majority have a 6-7 nucleotide match to the seed region of the siRNA. [22,23] This silencing of off-targets is undesirable, given the unpredictable consequences of altering gene activity. Due to the fact that siRNAs may act as micro RNAs (miRNAs) and silence multiple off-target genes simply by partial sequence complementarity, siRNAs must be meticulously designed and examined. [41]
Another mechanism by which unmodified siRNAs pose as toxicities is the stimulation of the innate immune response. Long dsRNA has been known to rapidly induce interferon responses by binding to dsRNA-activated protein kinase, 2',5'-oligoadenylate synthetase-RNase L system, or Toll-like receptors (TLRs), which are all mechanisms that serve in combating viral pathogens and other invaders. [24] A 2005 study demonstrated that certain siRNA sequence motifs induced TLR7-dependent stimulation of the immune response. [25] The use of longer siRNAs (>27 nucleotides long) must therefore be carefully balanced in consideration of the increased risk of immune stimulation.
Furthermore, siRNAs may compete with endogenous RNAs for miRNA processing pathways, leading to the saturation of miRNA pathways and ending in toxicity. [9] This has been observed in a study in which mice were given high doses of adeno-associated virus (AAV)-shRNA to the liver; the oversaturation of the RNA pathway resulted in fatality. [43]
Yet another challenge posed by siRNA is the safe and efficient delivery of siRNA to the desired organ and subsequently into target cells. Unless siRNAs are administered in large volumes, which cannot be performed on humans, siRNAs will be unable to penetrate tissue. [41] To resolve this issue, several approaches to delivering siRNAs into cells have been developed, including but not limited to lipid vesicles, viral vectors, and plasmids. [26,27,41]
Approaches to siRNA Delivery
The delivery of siRNAs to the appropriate cells is of utmost concern. Many publications have demonstrated that the systemic delivery of siRNAs to tissues successfully results in the knockdown of target mRNAs; this can be accomplished by the intravenous injection of siRNAs that have been packaged into liposomes or conjugated to a cholesterol group. [19] Liposomes, depicted in Figure 2, are the most commonly used carriers for delivery of various RNAi agents in vivo, as they can be easily prepared from biocompatible lipid or phospholipid ingredients and easily modified for specific purposes, such as the encapsulation of hydrophilic drug molecules. Cationic liposomes are especially convenient; the electrostatic interactions between the positively charged lipids and the negatively charged RNA molecules they encapsulate notably improve the efficiency of RNA encapsulation. [26] siRNAs encapsulated into cationic liposomes are taken up by the target cell through endocytosis; a further step to ensure that the desired cells are targeted is to incorporate ligands that bind to cell surface receptors on the targeted tissue into the cationic coat of the liposome. [41]
A liposome. A liposome is a spherical sac made up of a simple phospholipid membrane. siRNA may be efficiently delivered to target cells by the use of liposomes. (Adapted from [59])
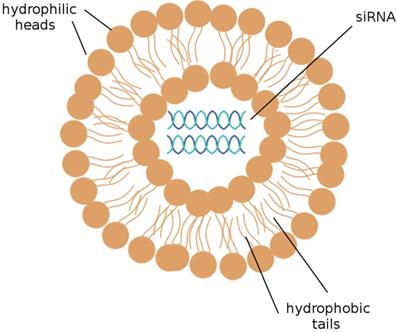
Viral vectors have also been designed to express siRNA within target cells. The advantage of using viral vectors is that siRNA can be introduced into non-dividing cells, such as neurons, but as with most viral approaches, the disadvantage of using viral vectors is the hassle of having to perform frequent, repeated administrations and the limited control over the types of cells that are transduced. [41] Using this approach, chemically synthesized siRNA may be encapsulated into vesicles derived from reconstituted viral envelopes. This reconstituted membrane vesicle would have to contain a protein from the original membrane of the virus that could facilitate binding to or fusion with the target cell membrane in order for the vesicle to be taken up by receptor-mediated endocytosis. [44]
Plasmid vectors carrying genes for the expression of either siRNA or shRNA may provide another alternative to introducing siRNA into a cell. Instead of encapsulating siRNA molecules themselves into a carrier, siRNA expression plasmids may be packaged and delivered to generate siRNAs within the cell. These siRNA plasmid vectors may be developed to generate highly efficient siRNAs that reliably assemble into RISCs and silence the target mRNA. [43] Plasmid vectors carrying DNA inserts for shRNA expression may be used to generate siRNA in a cell, as well; transcribed shRNA folds into a structure similar to a siRNA duplex, which can be processed by Dicer into units of siRNA. [27] By encapsulating these plasmids into viruses, the siRNA delivery process can be made highly efficient; the binding of the virus to the target cell surface, transduction, stability of the carrier, and protection against nucleases all appear to proceed smoothly and satisfactorily. [44]
Cationic cell penetrating peptides (CPPs) are also another option and have been developed to facilitate the intracellular delivery of molecules such as plasmid DNA, oligonucleotides, and siRNA into cells. The main advantage of using CPPs is that the chemical modification of siRNA is not required, allowing for the activity of the siRNA to be preserved and for the siRNA purification process to be more streamlined. [9] CPPs are comprised of short arginine strands and/or lysine-rich peptides. Because of their cationic nature, CPPs are able to efficiently accumulate within cells. However, since this approach to siRNA delivery requires the direct complexation of a CPP with the anionic phosphate backbone of siRNA, this efficiency is reduced. [46] Still, CPP-mediated siRNA delivery systems are able to enter cells either by endocytosis or by directly crossing the membrane. [9]
Therapeutic Applications of siRNA-Mediated RNAi on Breast Cancers
Given that siRNA has the ability to carry out sequence-specific gene silencing, recent interest has grown around applying siRNA-mediated RNAi gene therapy to breast cancer. Many genes associated with the development of breast cancer, including BRCA1, BRCA2, HER2, and p53, could serve as potential targets for this treatment. [27,33,49]
BRCA1 and BRCA2
The breast cancer susceptibility gene 1 (BRCA1) and breast cancer susceptibility gene 2 (BRCA2) are both associated with breast cancer, as well as ovarian cancer. [27] Mutations on the BRCA1 gene enhance the risk of breast cancer development by approximately 59% to 87%, whereas mutations on the BRCA2 gene enhance the risk of breast cancer development by approximately 38% to 80%. [29] Furthermore, the BRCA1 and BRCA2 genes both show an autosomal dominant manner of inheritance. An individual possessing a BRCA1 or BRCA2 germline pathogenic variant has a 50% chance of passing the mutated allele to his or her offspring. Not all individuals who possess a pathogenic variant of BRCA1 or BRCA2 have a parent affected by cancer, however; the variable age of cancer development, prophylactic surgery, and other factors play a role in this. [27]
BRCA1 and BRCA2 are tumor suppressor genes that play roles in pathways critical for controlling DNA damage, including double-strand break repair and transcription regulation. [28] BRCA1 is located at chromosome 17q21 and codes for a 1,863 amino-acid-long protein that functions in both checkpoint activation and gene regulation processes in response to DNA damage. [29, 47] The protein encoded by BRCA1 possesses two BRCA1 C-terminal (BRCT) domains at the C terminus and a RING (Really Interesting New Gene) finger domain at the N terminus, as shown in Figure 3. [30] The BRCT domain facilitates phospho-protein binding, whereas the RING domain has E3 ubiquitin ligase activity, which catalyzes protein ubiquitination. Mutations within the RING domain and the BRCT domain have both been observed in numerous inherited cancer-associated alleles of BRC1, indicating that both domains play a role in the suppression of breast cancer. [47]
BRCA1 and BRCA2 structure. BRCA1 possesses a RING domain at the N terminus and two BRCT domains at the C terminus. BRCA2 possesses eight BRC domains and DNA binding domains that bind double-stranded and single-stranded DNA. (Adapted from [56])
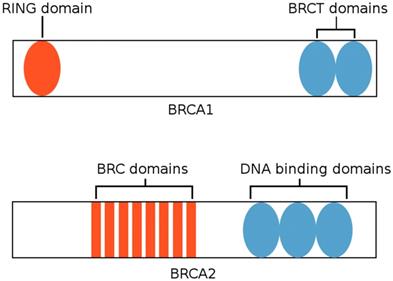
On the other hand, BRCA2 is located at chromosome 13q12.3, and the protein encoded by BRCA2 consists of 3,418 amino acids. [29] BRCA2 is primarily involved in the recruitment of the recombinase RAD51 to repair double-strand breaks. The protein encoded by BRCA2 possesses eight BRC repeats that bind RAD51 and a DNA-binding domain that binds both double-stranded DNA and single-stranded DNA. [47] This domain is comprised of five components: three oligonucleotide-binding folds (OB1, OB2, OB3), a 190 amino-acid-long α-helical domain, and a tower domain that protrudes from OB2. [48] Individuals with hereditary breast cancer or ovarian cancer exhibit point mutations within BRC repeats that compromise the interactions pertaining to RAD51. [47]
Although the use of siRNA alone may not serve as an effective treatment for BRCA1-positive breast cancers or BRCA2-positive breast cancers, siRNA-mediated silencing alongside other treatments can increase the potency of those other treatments. In a 2014 study, the expression of BRCA1 in HeLa cells was suppressed using siRNA-mediated knockdown. These BRCA1-suppressed cells became more sensitized to the proteasome inhibitors bortezomib and carfilzomib and ultimately initiated apoptosis, due to the fact that proteasome inhibitors prevent the degradation of pro-apoptotic factors. [30,45] siRNA could thus be applied to cells to increase the potency of proteasome inhibitors as well as other possible treatment agents. In a 2008 study, HeLa cells transfected with BRCA2-siRNA exhibited significantly lower levels of survival when exposed to radiation compared to HeLa cells transfected with negative-control siRNA or mock-siRNA; this opens up the possibility of using siRNA as a radiosensitizer in tumor radiotherapy. [40]
HER2
The human epidermal growth factor receptor 2 (HER2) gene, also known as HER2/neu, ERBB2, or CD340, is associated with poor breast cancer prognosis; the HER2 gene is amplified and the HER2 protein overexpressed in approximately 20% of all breast cancer cases. [31,33] As suggested by experimental evidence, the amplification of HER2 appears to lead to malignant transformation. [36] Breast cancer cells may possess from 25-50 copies of the HER2 gene and consequently a 40-100 times increase in the expression of HER2, ultimately culminating in up to 2 million HER2 proteins being expressed at the cell surface at a time. Unique characteristics of HER2 amplified breast cancers include resistance to hormonal agents, increased sensitivity to cytotoxic chemotherapeutic agents, and an increased tendency to metastasize to the brain. [35] Figure 4 depicts HER2 expression in a normal cell compared to the expression of HER2 in an overexpressing cell.
HER2 is located to chromosome 17, and HER2-positive breast cancers may possess either multiple copies of the HER2 gene with a normal chromosome 17 count or multiple copies of chromosome 17 with varying numbers of the HER2 gene on each chromosome. [39] HER2 encodes a receptor tyrosine kinase that initiates signal transduction pathways driving cell survival and proliferation in approximately 85% of breast cancers. [34] Similar to other HER family proteins, HER2 comprises an extracellular ligand binding domain, a transmembrane domain, and an intracellular tyrosine kinase domain. [35] However, because HER2 has no known ligand, it relies on either heterodimerization with another member of the HER family of proteins or homodimerization with another HER2 protein to be activated. [34] When overexpressed, HER2 tends to form heterodimers with other HER family proteins, resulting in powerful downstream signaling. [33]
HER2 expression. Cells overexpressing HER2 exhibit an abnormal number of HER2 proteins embedded in the membrane and an abnormal amount of HER2 mRNA expressed in the cell. The overexpression of HER2 may be due to an unusual number of HER2 gene copies on chromosome 17, as depicted here. (Adapted from [57])
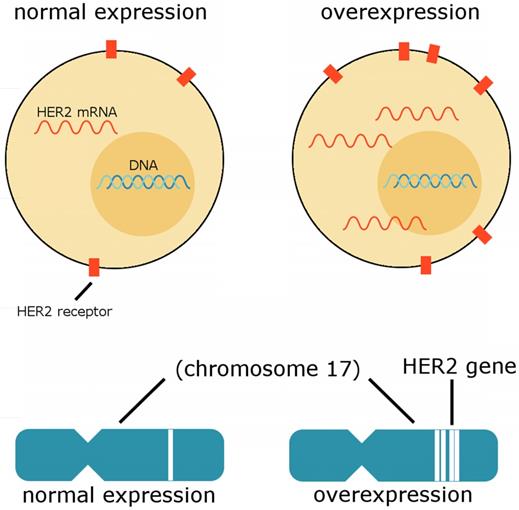
Structural alterations in the HER2 protein have been associated with resistance to trastuzumab, a HER2-targeted therapy. [33] A significant portion of patients have acquired resistance to HER2-targeted therapies, including trastuzumab and pertuzumab, leading to a combination of multiple drugs to be used in clinical treatments for HER2 amplified breast cancers. This, unfortunately, places a significant burden on patients. [31] siRNA may be able to overcome this barrier; a study in 2004 demonstrated that the siRNA-mediated silencing of the HER2 gene induces apoptosis in breast cancer cells overexpressing HER2, inhibiting the proliferation of these breast cancer cells. [36] However, another challenge that remains is that safe and effective methods of delivering siRNA in vivo have yet to be approved. Viral-based siRNA and shRNA strategies have been found effective, but significant issues concerning insertional mutagenesis and immunogenic response remain. [31] Non-viral-based modes of siRNA delivery have been developed and appear to be more viable; a publication from 2011, for instance, demonstrated that the use of a poly (β-L-malic acid) (PMLA)-based nanobiopolymer conjugated with trastuzumab and HER2 antisense inhibited the growth of HER2-positive human breast tumors in xenogeneic mouse models. [37] Another publication from 2012 also demonstrated that the delivery of Polo-like kinase 1 (PLK1)-siRNA using fusion proteins of single-chain fragmented antibodies (ScFvs) suppressed HER2-positive breast cancer growth and metastasis. [38] The only concern, however, is that these studies used BT474 derived tumors, which are vulnerable to trastuzumab, so other complications regarding efficacy may arise with the application of the aforementioned treatments in trastuzumab-resistant cancer cells.
p53
The tumor protein p53 (p53) gene, also known as TP53, is a tumor suppressor gene commonly associated with breast cancer, as well as a myriad of other cancers. The mutational inactivation of p53 occurs in approximately 50% of human cancers, making it one of the most common and well-known cancer-associated genes. [49]
The p53 protein is heavily involved in the stress response of human cells; p53 responds to a myriad of stress signals, including signals originating from DNA damage, the deregulated expression of oncogenes, telomere erosion, and metabolic deprivation. [50] Although the p53 gene is normally highly regulated, its activation can be triggered by posttranslational modifications to the p53 protein, such as ubiquination, phosphorylation, acetylation, and methylation. [49] Depending on the type of stress signals being received, activated p53 may respond by inducing apoptosis, senescence, cell-cycle arrest, DNA repair, or autophagy—all of these being anticancer mechanisms. [49, 51] The p53 monomer possesses two DNA binding domains; one is responsible for binding to sequence-specific DNA response elements that are located near the promoters of the target genes of p53, while the other binding domain, localized to the C-terminus, allows for p53 to non-specifically bind to DNA. [53]
Under unstressed, normal conditions, p53 is practically undetectable due to its extremely short half-life. This instability of p53 can be attributed to MDM2, an E3 ubiquitin ligase, which acts as the negative regulator of p53 and targets p53 for proteasome-mediated degradation. [49] MDMX also serves as a negative regulator of p53, working to block the transcriptional activity of p53. However, because p53 also stimulates the expression of MDM2 and MDMX, the three genes are in a constant feedback loop, regulating the expression of one another. [52] Another source of instability stems from the properties of the p53 protein itself—the melting temperature of p53 is only slightly above body temperature. [49] Most tumor suppressors require the loss of function in both alleles in order for tumorigenesis to occur, but mutation in only one allele of p53 results in the loss of its function and the development of tumors. [53]
Recently, siRNA-mediated RNAi has been studied as a treatment against mutant p53 in triple-negative breast cancer cells, which are resistant to conventional cancer therapy. A study conducted in 2015 revealed that therapy involving the use of p53-siRNA and epigallocatechingallate (EGCG), the most abundant compound found in green tea, successfully silenced the mutant p53 gene and resulted in the activation of pro-apoptotic genes, as well as the inhibition of pro-survival genes, cell network formation, and autophagy. [54] A study conducted in 2016 also showed that apoptosis is induced following the knockdown of mutant p53 and tumor necrosis factor (TNF) using siRNA. [55] These findings strongly suggest that siRNA could be an extremely effective agent for the treatment of both aggressive breast cancer phenotypes such as triple-negative breast cancer and milder phenotypes of breast cancer.
Conclusion
Breast cancer continues to plague victims worldwide, but siRNA-mediated RNAi provides hope as a future potential treatment. The potential targets of breast cancer to which siRNA-mediated RNAi may be applicable are not limited to BRCA1, BRCA2, HER2, and p53; theoretically, siRNA could be synthesized to target any specific gene, opening up a plethora of opportunities.
siRNA thus holds incredibly promising potential as a form of gene therapy for not only breast cancer but possibly other cancers and diseases as well. Although its limitations may pose challenges, current efforts to modify siRNA and develop more safe and efficient modes of its delivery to target cells seem to be advancing RNAi technology in the right direction.
Competing Interests
The authors have declared that no competing interest exists.
References
1. DeSantis CE, Bray F, Ferlay J, Lortet-Tieulent J, Anderson BO, Jemal A. International Variation in Female Breast Cancer Incidence and Mortality Rates. Cancer Epidemiology, Biomarkers, & Prevention. 2015;24:1495-506
2. Dieterich M, Stubert J, Reimer T, Erickson N, Berling A. Influence of Lifestyle Factors on Breast Cancer Risk. Breast Care. 2014;9:407-14
3. Shah R, Rosso K, Nathanson SD. Pathogenesis, prevention, diagnosis and treatment of breast cancer. World Journal of Clinical Oncology. 2014;5:283-98
4. Collaborative Group on Hormonal Factors in Breast Cancer. Familial breast cancer: collaborative reanalysis of individual data from 52 epidemiological studies including 58,209 women with breast cancer and 101,986 women without the disease. Lancet. 2001;358:1389-99
5. Tokatlian T, Segura T. siRNA applications in nanomedicine. Wiley Interdisciplinary Reviews. Nanomedicine and Nanobiotechnology. 2010;2:305-15
6. Carthew RW, Sontheimer EJ. Origins and Mechanisms of miRNAs and siRNAs. Cell. 2009;136:642-655
7. Zhang H, Kolb FA, Jaskiewicz L, Westhof E, Filipowicz W. Single processing center models for human Dicer and bacterial RNase III. Cell. 2004;118:57-68
8. Kim DH, Rossi JJ. RNAi mechanisms and applications. BioTechniques. 2008;44:613-616
9. Wang J, Lu Z, Wientjes MG, Au JL-S. Delivery of siRNA Therapeutics: Barriers and Carriers. The AAPS Journal. 2010;12:492-503
10. Kanellopoulou C, Muljo SA, Kung AL, Ganesan S, Drapkin R, Jenuwein T. et al. Dicer-deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes & Development. 2005;19:489-501
11. Agrawal N, Dasaradhi PVN, Mohmmed A, Malhotra P, Bhatnagar RK, Mukherjee SK. RNA Interference: Biology, Mechanism, and Applications. Microbiology and Molecular Biology Reviews. 2003;67:657-85
12. Matranga C, Tomari Y, Shin C, Bartel DP, Zamore PD. Passenger-strand cleavage facilitates assembly of siRNA into Ago2-containing RNAi enzyme complexes. Cell. 2005;123:607-20
13. Elkayam E, Kuhn C-D, Tocilj A, Haase AD, Greene EM, Hannon GJ. et al. The Structure of Human Argonaute-2 in Complex with miR-20a. Cell. 2012;150:100-10
14. Bertrand JR, Pottier M, Vekris A, Opolon P, Maksimenko A, Malvy C. Comparison of antisense oligonucleotides and siRNAs in cell culture and in vivo. Biochem Biophys Res Commun. 2002;296:1000-4
15. Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806-11
16. Orban TI, Izaurralde E. Decay of mRNAs targeted by RISC requires XRN1, the Ski complex, and the exosome. RNA. 2005;11:459-69
17. Shen B, Goodman HM. Uridine addition after microRNA-directed cleavage. Science. 2004;306:997
18. Gredell JA, Berger AK, Walton SP. Impact of target mRNA structure on siRNA silencing efficiency: a large-scale study. Biotechnology and Bioengineering. 2008;100:744-55
19. Kim DH, Rossi JJ. RNAi mechanisms and applications. BioTechniques. 2008;44:613-6
20. Miele E, Spinelli GP, Miele E, Di Fabrizio E, Ferretti E, Tomao S. et al. Nanoparticle-based delivery of small interfering RNA: challenges for cancer therapy. International Journal of Nanomedicine. 2012;7:3637-57
21. Jackson AL, Bartz SR, Schelter J, Kobayashi SV, Burchard J, Mao M. et al. Expression profiling reveals off-target gene regulation by RNAi. Nature Biotechnology. 2003;21:635-7
22. Lin X, Ruan X, Anderson MG, McDowell JA, Kroeger PE, Fesik SW. et al. siRNA-mediated off-target gene silencing triggered by a 7 nt complementation. Nucleic Acids Research. 2005;33:4527-35
23. Jackson AL, Burchard J, Schelter J, Chau BN, Cleary M, Lim L. et al. Widespread siRNA “off-target” transcript silencing mediated by seed region sequence complementarity. RNA. 2006;12:1179-87
24. Aagaard L, Rossi JJ. RNAi Therapeutics: Principles, Prospects and Challenges. Advanced Drug Delivery Reviews. 2007;59:75-86
25. Hornung V, Guenthner-Biller M, Bourquin C, Ablasser A, Schlee M, Uematsu S. et al. Sequence-specific potent induction of IFN-alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nature Medicine. 2005;11:263-70
26. Xue HY, Guo P, Wen W-C, Wong HL. Lipid-Based Nanocarriers for RNA Delivery. Current Pharmaceutical Design. 2015;21:3140-7
27. Petrucelli N, Daly MB, Pal T. BRCA1- and BRCA2-Associated Hereditary Breast and Ovarian Cancer. Adam MP, Ardinger HH, Pagon RA. et al (eds). GeneReviews® [Internet]. Seattle: University of Washington, Seattle. 1993
28. Gudmundsdottir K, Ashworth A. The roles of BRCA1 and BRCA2 and associated proteins in the maintenance of genomic stability. Oncogene. 2006;25:5864-74
29. Karami F, Mehdipour P. A Comprehensive Focus on Global Spectrum of BRCA1 and BRCA2 Mutations in Breast Cancer. BioMed Research International. 2013;2013:928562
30. Gu Y, Bouwman P, Greco D, Saarela J, Yadav B, Jonkers J. et al. Suppression of BRCA1 sensitizes cells to proteasome inhibitors. Cell Death & Disease. 2014;5:e1580
31. Gu S, Hu Z, Ngamcherdtrakul W, Castro DJ, Morry J, Reda MM. et al. Therapeutic siRNA for drug-resistant HER2-positive breast cancer. Oncotarget. 2016;7:14727-41
32. Prat A, Carey LA, Adamo B, Vidal M, Tabernero J, Cortés J. et al. Molecular Features and Survival Outcomes of the Intrinsic Subtypes Within HER2-Positive Breast Cancer. JNCI Journal of the National Cancer Institute. 2014;106:dju152
33. Diver EJ, Foster R, Rueda BR, Growdon WB. The Therapeutic Challenge of Targeting HER2 in Endometrial Cancer. The Oncologist. 2015;20:1058-68
34. Gutierrez C, Schiff R. HER 2: Biology, Detection, and Clinical Implications. Archives of Pathology & Laboratory Medicine. 2011;135:55-62
35. Moasser MM. The oncogene HER2; Its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene. 2007;26:6469-87
36. Faltus T, Yuan J, Zimmer B, Krämer A, Loibl S, Kaufmann M. et al. Silencing of the HER2/neu Gene by siRNA Inhibits Proliferation and Induces Apoptosis in HER2/neu-Overexpressing Breast Cancer Cells. Neoplasia. 2004;6:786-95
37. Inoue S, Ding H, Portilla-Arias J, Hu J, Konda B, Fujita M. et al. Polymalic Acid-Based Nanobiopolymer Provides Efficient Systemic Breast Cancer Treatment by Inhibiting both HER2/neu Receptor Synthesis and Activity. Cancer Research. 2011;71:1454-64
38. Yao YD, Sun TM, Huang SY, Dou S, Lin L, Chen JN. et al. Targeted delivery of PLK1-siRNA by ScFv suppresses Her2+ breast cancer growth and metastasis. Science Translational Medicine. 2012;4:130ra48
39. Perez EA, Reinholz MM, Hillman DW, Tenner KS, Schroeder MJ, Davidson NE. et al. HER2 and Chromosome 17 Effect on Patient Outcome in the N9831 Adjuvant Trastuzumab Trial. Journal of Clinical Oncology. 2010;28:4307-15
40. Yu D, Sekine E, Fujimori A, Ochiya T, Okayasu R. Down regulation of BRCA2 causes radio-sensitization of human tumor cells in vitro and in vivo. Cancer Science. 2008;99:810-5
41. Felekkis K, Deltas C. RNA Intereference: A powerful laboratory tool and its therapeutic implications. Hippokratia. 2006;10:112-5
42. Peralta-Zaragoza O, De-la-O-Gómez F, Deas J, Fernández-Tilapa G, Fierros-Zárate G, Gómez-Cerón C. et al. Selective silencing of gene target expression by siRNA expression plasmids in human cervical cancer cells. Methods of Molecular Biology. 2015;1249:153-71
43. Grimm D, Streetz KL, Jopling CL, Storm TA, Pandey K, Davis CR. et al. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature. 2006;441:537-41
44. Oliveira S, Storm G, Schiffelers RM. Targeted Delivery of siRNA. Journal of Biomedicine and Biotechnology. 2006;2006:63675
45. Crawford LJ, Walker B, Irvine AE. Proteasome inhibitors in cancer therapy. Journal of Cell Communication and Signaling. 2011;5:101-10
46. Mo RH, Zaro JL, Shen W-C. Comparison of Cationic and Amphipathic Cell Penetrating Peptides for siRNA Delivery and Efficacy. Molecular Pharmaceutics. 2012;9:299-309
47. Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nature Reviews Cancer. 2012;12:68-78
48. Yang H, Jeffrey PD, Miller J, Kinnucan E, Sun Y, Thoma NH. et al. BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science. 2002;297:1837-48
49. Wang Z, Sun Y. Targeting p53 for Novel Anticancer Therapy. Translational Oncology. 2010;3:1-12
50. Walerych D, Napoli M, Collavin L, Del Sal G. The rebel angel: mutant p53 as the driving oncogene in breast cancer. Carcinogenesis. 2012;33:2007-17
51. Zilfou JT, Lowe SW. Tumor Suppressive Functions of p53. Cold Spring Harbor Perspectives in Biology. 2009;1:a001883
52. Zhang Q, Zeng SX, Lu H. Targeting p53-MDM2-MDMX Loop for Cancer Therapy. Sub-Cellular Biochemistry. 2014;85:281-319
53. Aramayo R, Sherman MB, Brownless K, Lurz R, Okorokov AL, Orlova EV. Quaternary structure of the specific p53-DNA complex reveals the mechanism of p53 mutant dominance. Nucleic Acids Research. 2011;39:8960-71
54. Braicu C, Pileczki V, Pop L, Petric RC, Chira S, Pointiere E. et al. Dual Targeted Therapy with p53 siRNA and Epigallocatechingallate in a Triple Negative Breast Cancer Cell Model. PLOS ONE. 2015;10:e0120936
55. Pileczki V, Pop L, Braicu C, Budisan L, Bolba Morar G, del C Monroig-Bosque P, Berindan-Neagoe I. Double gene siRNA knockdown of mutant p53 and TNF induces apoptosis in triple-negative breast cancer cells. OncoTargets and Therapy. 2016;9:6921-33
56. Roy R, Chun J, Powell SN. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nature Reviews Cancer. 2011;12:68-78
57. Lin F, Baracat E, Carvalho F. Amplification of the Oncogene HER2 in Breast Cancer: Molecular Basis and Therapeutic Significance. Journal of the Senologic International Society. 2012:1
58. Arenz C, Schepers U. RNA interference: From an ancient mechanism to a state of the art therapeutic application? Die Naturwissenschaften. 2003;90:345-59
59. Yu-Wai-Man C, Tagalakis A, Manunta M, Hart S, Khaw P. Receptor-targeted liposome-peptide-siRNA nanoparticles represent an efficient delivery system for MRTF silencing in conjunctival fibrosis. Nature Scientific Reports. 2016;6:21881
Author contact
![]() Corresponding author: Yi Lu, Ph.D., Department of Pathology and Laboratory Medicine, University of Tennessee Health Science Center, Cancer Research Building, Room 258, 19 South Manassas Street, Memphis, TN 38163 (USA). Tel.: (901) 448-5436; Fax.: (901) 448-5496; E-mail: yluedu
Corresponding author: Yi Lu, Ph.D., Department of Pathology and Laboratory Medicine, University of Tennessee Health Science Center, Cancer Research Building, Room 258, 19 South Manassas Street, Memphis, TN 38163 (USA). Tel.: (901) 448-5436; Fax.: (901) 448-5496; E-mail: yluedu