Oncomedicine 2019; 4:17-26. doi:10.7150/oncm.27020 This volume Cite
Review
The Functional Role of Bcl-2 Family of Proteins in the Immune System and Cancer
Aileen Chen1, Chikezie O. Madu2, Yi Lu3 ![]()
1. Department of Biology and Advanced Placement Biology, White Station High School, Memphis, Tennessee 38117. USA.
2. Departments of Biology and Advanced Placement Biology, White Station High School, Memphis, Tennessee 38117. USA.
3. Departments of Pathology and Laboratory Medicine, University of Tennessee Health Science Center, Memphis, TN 38163, USA.
Received 2018-5-2; Accepted 2018-7-13; Published 2019-6-14
Abstract

The Bcl-2 family of proteins plays a significant role in regulating the cell cycle. It plays a crucial role in regulating homeostasis by helping to maintain proper cell number and eliminating potentially malignant cells. The cell achieves the delicate balance through apoptosis or programmed cell death. The Bcl proteins are involved in the intrinsic apoptotic pathway, which is especially important in the development of the immune system. Apoptosis is involved in proper positive and negative selection during the development of B- and T-cells. The detection of any gene of the Bcl-2 family often results in the abnormal development of lymphocytes. The balance of pro-apoptotic proteins versus anti-apoptotic proteins, which operate at the organelle level, determines if the lymphocyte proliferates normally or abnormally. A critical tumor suppressor is the ability of cells to self-disrupt and undergoes cell death through apoptosis. Cooperation between cells is essential; without the ability to respond to external stimuli, cells lose the ability to respond correctly to developmental cues. Cells that evade apoptosis have a greater potential to become malignant because they are unregulated and do not respond correctly to external signals. The overexpression of the pro-survival (anti-apoptotic) proteins of the Bcl-2 family induces the cell to not respond to an external signal, prolonging cell survival and increasing the chance of becoming malignant.
Keywords: bel-2, apoptosis, anti-apoptotic protein, cancer
Introduction
An essential component of regulating organismal homeostasis is maintaining the proper number of cells and eliminating damaged or malignant cells. Apoptosis, or programmed cell death, is the mechanism responsible for this balance. The term “apoptosis” was first coined by Kerr et al. [57] to differentiate a type of cell death from necrosis. Apoptosis is linked to cell shrinkage and DNA fragmentation, where the membrane blebs and yields to small vesicles, which are then engulfed and digested by neighboring cells. [1] Apoptosis is crucial for proper positive and negative selection during the B and T cell development. [1] The intrinsic apoptosis pathway (via mitochondria) is activated in response to internal cellular stress and is primarily regulated by the Bcl-2 family of proteins.
The 25 currently identified members of the Bcl-2 family of proteins are localized to the mitochondria, smooth endoplasmic reticulum, and perinuclear membranes in hematopoietic cells. [9] The outer mitochondrial membrane permeabilization is considered the “point of no return” from apoptotic cell death, triggering the release of proteins that mediate cell death, such as cytochrome c, into the cytoplasm of the cell. [7] The permeabilization of the outer membrane is mediated by certain Bcl-2 family members that regulate apoptosis among a collection of pro- and anti-apoptotic proteins [8].
The Bcl-2 gene was first discovered as a part of a chromosomal translocation in B-cell lymphoma. [2] Bcl-2 was implicated in cancer through its involvement in the t14; 18 chromosome translocation. [50] It is known as the first oncogene to inhibit cell death opposed to promoting cell proliferation. [3] The Bcl-2 family of proteins plays a central role in the regulation of cell death and is involved in regulating cell death mechanisms including apoptosis, necrosis, and autophagy. [4] The anti-apoptotic members of this family, such as the Bcl-SL and the Bcl-W, prevent apoptosis by either preventing the release of mitochondrial apoptogenic factors or sequestering proforms of death-driving cysteine proteases called caspases [6].
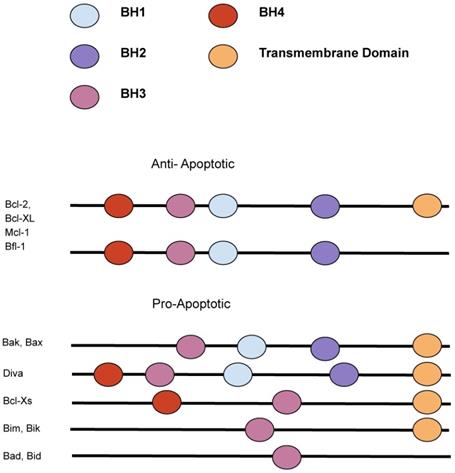
Changes to the Bcl-2 gene structure or copy number are estimated to occur in as many as over half of human cancers. [4] The Bcl-2 protein family shares one to four highly conserved regions in structure and sequence called the Bcl-2 homology (BH) domains. Based on the domains, they are divided into three functional subgroups, the multidomain anti-apoptotic (Bcl-2, Bcl-XL, Bcl-W, Mcl-1, BFL-1), the multidomain pro-apoptotic (BAK, BAX, BOK), and the BH3-only proteins (BIM, BID, BAD, NOXA, PUMA, BMF, BIK, HRK). [1] The BH3- only proteins are named so because they only share the third BH domain with the other Bcl-2 family proteins, acting as cellular sentinels that in the time of stress can bind discrete multidomain Bcl-2 proteins and initiate apoptosis. [5] The anti-apoptotic subfamily suppresses apoptosis and contains all four of the Bcl-2 homology domains, while some of the pro-apoptotic proteins contain Bcl-2 homology domains 1-3, known as the “multidomain proteins.” [9] The delicate balance of pro-apoptotic versus anti-apoptotic proteins of the Bcl-2 family of proteins, which operate at the organelle level, determines if the lymphocyte lives or dies.
An essential tumor suppressor is the ability of cells to self-disrupt and undergoes cell death through apoptosis. The cooperation between cells is critical, without the ability to respond to external stimuli, cells lose the ability to respond correctly to developmental cues. Cells that evade apoptosis have a greater potential to become malignant because they are unregulated by neighboring cells and do not respond correctly to external signals, hence why they often do not undergo apoptosis when they are supposed to.
The Function of the Bcl-2 family
The Bcl-2 family, consisting of both proapoptotic and antiapoptotic genes, regulates apoptosis, upholding the balance between cell death and proliferation. Deletion of potentially malignant cells, usually through apoptosis, protects against a variety of diseases, including autoimmunity and cancer. [47] Apoptosis is triggered through the extrinsic pathway, through ligation, or the intrinsic (also called the mitochondrial or Bcl-2 regulated) pathway, which is induced by developmental cues, deprivation of growth factors, or other cytotoxic stimuli. The intrinsic pathway is regulated by the Bcl-2 family. [47] The Bcl-2 family, consisting of both apoptotic and anti-apoptotic genes, is divided into three functional subgroups: the multidomain anti-apoptotic (Bcl-2, Mcl-1, Bcl-W, BFL-1, Bcl-XL), the multidomain pro-apoptotic proteins (BAK, BAX, BOK) and BH3-only (BMF, BID, BIM, HRK, NOXA, PUMA, BAD, BIK). [1,10] (Figure 1) The BAK, BAX, and BOK lead to the formation of mitochondrial pores that increase the permeability of the mitochondrial membrane that releases cytochrome c into the cytosol leading to cell death. [10,13] The pro- and anti-apoptotic proteins have a similar C-terminal membrane localization point, similar three-dimensional structures, and three or four Bcl-2 homology domains (BH1, BH2, BH3, and BH4). The slight structural differences caused by a few amino acids determine the opposing roles of the proapoptotic and antiapoptotic proteins. The BH3-only proteins are pro-apoptotic proteins that lack all the Bcl-3 homology domains except for BH3. The pro-apoptotic proteins contain BH1 to BH3, while the pro-survival proteins contain all four Bcl-2 homology domains. The BH3-only proteins regulate the other members of the Bcl-2 family proteins to promote apoptosis by binding to their BH3 domain. [10,11] In response to cellular signals, such as DNA damage or oncogene activation, BH3-only members are activated by transcription of the gene or posttranslational modification of the mRNA [13].
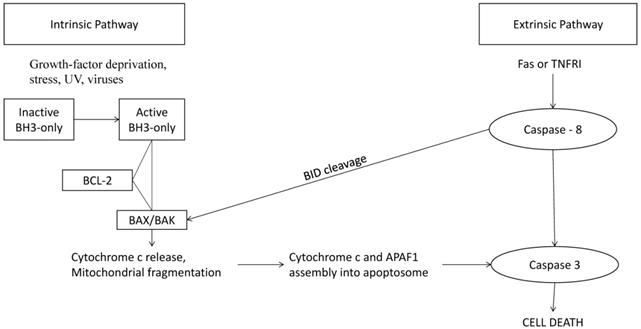
Apoptosis in the cell can be divided into three stages: initiation, regulation, and execution. In initiation, the cells undergo DNA damage or stress prompting a signal cascade through either an extrinsic or intrinsic pathway. In the second phase, the regulatory phase, these signals combine and decide whether or not the cell will undergo apoptosis. In the execution phase, the caspases are cleaved, and the cell is engulfed by phagocytic cells. [10,16] The intrinsic (also known as the “stress” or “mitochondrial”) [48] pathway is regulated by the Bcl-2 family of pro-apoptotic and anti-apoptotic proteins. (Figure 2) Certain members of the BH3 family initiate oligomerization of BAK, leading to mitochondrial outer membrane permeabilization and permitting the release of pro-apoptotic factors, such as cytochrome c, into the cytoplasm. [12,14] Cytochrome c, along with APAF-2 and caspase-9, forms the apoptosome, a holoenzyme that cleaves and activates caspase-3, leading to widespread proteolysis and cell death. [13,15] The extrinsic pathway, however, is triggered by the binding of ligands to proteins on the cell surface. [17] These receptors are members of the tumor necrosis factor family (TNF) that contains an intracellular death domain that activates caspase-8 through an adapter protein called Fas-associated death domain (FADD) at the cell surface. [10] Mostly, the interaction between the pro- and anti-apoptotic proteins through their BH3 domains determines the cell fate [10].
The Bcl-2 family members. The Bcl-2 family can be divided into proapoptotic and anti-apoptotic categories. [90]
The intrinsic and extrinsic pathway that lead to cell death. The Bcl-2 family has strategic checkpoints throughout both the intrinsic and extrinsic pathway that could control whether the cell undergoes apoptosis. [11]
Apoptosis is essential for embryogenesis, tissue homeostasis, and defense against pathogens. The deregulation of the Bcl-2 family can lead to immune diseases and even cancer. [49] Apoptosis is essential in embryogenesis to form the different structures of the human body. In tissue homeostasis, apoptosis is vital because it regulates the number of cells and results in the death of malignant cells. Apoptosis also defends against pathogens by inducing cell death in infected, damaged cells. If the ability to undergo apoptosis is disabled within the cell, the likelihood of the cell to become a malignant cell increases.
Overexpression of any members of the Bcl-2 prosurvival family can protect the mutant cell from a range of external stimuli; however, the underexpression of any of the Bcl-2 prosurvival proteins have direct consequences in normal tissue development and homeostasis. [11] For example, Bcl-2 is critical for the survival of the renal epithelial stem cells in embryogenesis and mature B cell and T cell lymphocytes. Bcl-w has a selective role in spermatogenesis while Bcl-xL is involved in the survival of erythroid progenitors and neuronal cells in embryogenesis. [56] Mcl-1 deficient mice have shown that Mcl-1 is vital for implantation in early development, survival of hematopoietic stem cells and mature B and T lymphocytes. [11] The overexpression of proteins in the Bcl-2 family can result in prolonged cell survival, which could induce the formation of tumors or cancer.
The special position of the BH3-only proteins acts as the fulcrum to determine whether to tip the scales for cell death or cell proliferation. [60] However, the mechanisms that lead to the activation of the BH3-only proteins differ between each member of the BH3 subfamily along with different apoptotic stimuli [60].
Role of Bcl-2 Proteins in the Immune System
The proliferation of T lymphocytes is highly regulated by the Bcl-2 family. Apoptosis is involved in proper positive and negative selection during the development of B cells, plasma cells, and T lymphocytes. (Table 1) The deletion of any protein of the Bcl-2 family results in the abnormal development of lymphocytes which could result in lymphocytes proliferating prematurely or containing mutations that could cause autoimmune disorders. [1] The premature proliferation is often the result upsetting the delicate balance between the pro-apoptotic and anti-apoptotic proteins, which may result in autoimmune disorders or immunosuppressives. BH3-only members of the Bcl-2 family, which includes BIM, BMF, BIK, BAD, BID, PUMA, NOXA, and HRK, are especially essential to the immune system. [41] They have essential roles in development, homeostasis, immunity, and tumor suppression [37].
Specific anti-apoptotic Bcl-2 family proteins and their presence in specific immune cells.
| BFL-1 | Neutrophil |
| Mcl 1 | Hematopoietic Stem Cell |
| Regulatory T Cell | |
| Pro B Cell | |
| Pre B Cell | |
| Double Negative T cell | |
| BCL-2 | Activated T Cell |
| Double Negative T cell | |
| BCL-XL | Double Positive T Cell |
The different reliance of the various immune cells on unique Bcl-2 proteins allow for target drugs for specific immune sets to be created. [1]
Anti-apoptotic
Mcl-1, localized to intracellular membranes, especially the mitochondrial membrane, [1] is widely expressed in human and murine tissues and has a noticeable presence in many human tumors. [23] Though not as potent as Bcl-2, Mcl-1 has been shown to enhance cell survival and delay cell death. [24] The Mcl-1 protein also plays an essential role in human immune maintenance. The disruption of the Mcl-2 proteins resulted in embryonic lethality, indicating that the Mcl-1 gene is essential for preimplantation development and implantation. [22] These models quickly develop incredibly reduced numbers of hematopoietic stem cells. [1], suggesting that the Mcl-1 protein has a function beyond regulating apoptosis. [22] Mcl-1 is essential for the survival of plasma cells and germinal center formation. [28] Inducible deletion of the Mcl-1 in mice results in the ablation of the bone marrow, leading to the loss of early bone marrow progenitor populations. [25] Mcl-1 deletions in T cells leads to a reduction in the number of T lymphocytes, due to a blockage at the DN2/3 stage of development in the thymus. [26,27] Mcl-1 is critical for the survival of regulatory T (Treg) cells, a crucial immunosuppressive. [27] Without Mcl-1, the mice experienced weight loss and death within 4-8 weeks [27].
The overexpression of the anti-apoptotic protein, Bcl-2, with dysregulated c-my encourages the proliferation of immature B cell and tumorigenesis. [1] The expression of Bcl-2 alone, however, sustains the cell's survival without increasing the cell proliferation. [20] Ultimately, the expression of Bcl-2 leads to increased numbers of plasma cells, pre-B cells, and T cells that demonstrate increased longevity in cultures. [1] However, the deletion of the Bcl-2 gene leads to a decrease in the number of double-positive (DP) thymocytes and peripheral T and B cells.The thymocytes with a Bcl-2 deletion often are more susceptible to apoptotic stimuli [21].
Loss of the Bcl-XL protein has a minimal effect the overall survival of T cells. However, specific deletion of the Bcl-XL gene in B cells expressing activation-induced cytidine deaminase leads to a significant decrease of antigen-specific plasma cells in the bone marrow in the days directly following immunization, but after 21 days, the number normalizes. [29] This suggests that Bcl-XL assists the survival of newly generated plasma cells. Mice lacking Bcl-XL have reduced DP thymocytes, but they have a normal number of peripheral lymphocytes, implying that Bcl-XL is not alone in lymphocyte homeostasis [30].
Pro-apoptotic
Though the pro-apoptotic proteins BAK and BAX play redundant roles in the initiation of MOMP, the deletion of either or both of them leads to apoptotic defects. The deletion of both the BAK and BAX proteins leads to high embryonic lethality with high rates of developmental and neuronal defects in the resulting mice. The BAX/BAK-deficient mice have an increase in both lymphoid and myeloid cells, leading to the enlargement of the primary lymphoid organs and lymphocyte infiltration into peripheral organs. [17] The lymphocytes of the deficient mice are resistant to the activators of the intrinsic apoptotic pathway. These mice with T cell-specific BAX/BAK knockouts have abnormal thymocyte development and an increased buildup of double-negative cells in the thymus. [18] Deficient animals have thymocytes that are resistant to apoptosis, and those deficient animals develop T-cell lymphoma with average ten-month survival time [27].
BH3-Only Proteins
Deletions in BIM, considered to be the master regulator of immune cell homeostasis, can result in an increased number of lymphoid and myeloid cells and defects in thymocyte development. [1,31] In reduced levels of BIM, CD4+ T cells become both longer-lived and functionally impaired. [32] BIM is important for negative selection of immature T cells in the thymus and plays an essential role in regulating the survival of the cell. Decreasing BIM expression leads to prolonged survival of the CD4+ T cells that are susceptible to defects and become progressively unable to defend against pathogens. [32] BIM-deficient mice are unable to eliminate reactive thymocytes. [46] Deletion of additional BH3-only proteins facilitates immune dysfunction seen in BIM knockout models. [1] The additional deletion of PUMA, combined with the deletion of BIM, increases resistance to apoptotic stimuli. The combined deletion of BIM, PUMA, and BID leads to apoptotic resistance that has not been characterized in the model mice [33].
PUMA, p53 upregulated modulator of apoptosis, [13] is tightly regulated by the tumor suppressor p53. [35] Resultant sequences of genomic DNA revealed that the transcription of PUMA, located on chromosome 19, contains four exons, with the initial start codon in exon 2. [35] PUMA has been implicated as a mediator of apoptosis in immune cells. BIM and PUMA target pro-survival proteins and are much more potent killers than other BH3-only proteins. [37] PUMA works with BIM to regulate activated T cell action following an immune response. [1] During cellular stress, p53 blocks the progression of the cell cycle, allowing time for DNA repair. [40] More than 50% of PUMA deficient mice succumbed by 48 hours and 84% within 71 hours. [40] Deletion of PUMA leads to apoptotic resistance and an impaired regulation of neutrophil contractions after an immune response [39].
BID, a BH3 only pro-apoptotic member, binds to either Bcl-2 or BAX to promote apoptosis. [38] BID magnifies the death receptor signals received, connects the extrinsic and intrinsic pathways, and triggers the mitochondrial apoptosis pathway. [42] BID deficient mice undergo normal development and display balanced homeostasis at birth, but when tested with pathologic Fas activation, BID-deficient livers proved to be resistant to Fas-mediated apoptosis. [42] Aging BID-deficient mice exhibited an elevated absolute neutrophil count at 18-24 months accompanied by mild hepatosplenomegaly due to the infiltration of myeloid cells. However, in younger populations (less than 12 months old), the hematopoietic cell populations were indistinguishable from age from wild-type controls. [42] A detailed analysis of the bone marrow of the young BID-deficient mice demonstrated increased colony-forming units of macrophages. [42] This appears to have potentially uncovered a defect that is compensated for in vivo in the young mice, but the defect appears to emerge with age. [42] BID also may play a vital role in the survival of Langerhans cells, a subset of dendritic cells, because BID-deficient Langerhans cells are more resistant to CD4+ T-cell-mediated apoptosis [43].
NOXA, another BH3 member regulated by p53, plays a minor role in regulating the development and maintenance of effector memory T cells during and following an immune response. NOXA deficient mice result in memory cell inflation, but no obvious pathology. However, in continuous high-level T cell activation, the reduced apoptosis of T cells quickly led to severe organ pathology and premature death of the NOXA deficient mice. [44] The results demonstrated NOXA as an essential regulator of the number of memory cells formed during the infection, which could lead to excessive accumulation of primed cells that could result in severe pathology [44].
Cells lacking BAD exhibit normal B and T cell development and BAD-deficient animals have normal sensitivity to apoptotic stimuli. [46] BAD seems to be most important in B cell ontogeny and maturation; BAD-deficient mice have reduced IgG production after the stimulation of lipopolysaccharide and older mice develop diffuse large B cell lymphoma that progressively worsens after ionizing radiation [46]. BMF deficient mice maintain normal T cell number and exhibit no abnormalities in thymocyte development, but BMF deficient mice do experience B-cell hyperplasia. Deficiency in B and T cells confers resistance to apoptosis in response to HDAC inhibition or glucocorticoids. [47] BMF deficiency quickens the development of γ irradiation thymic lymphomas, indicating that BMF plays a critical role in apoptosis and functions as a tumor suppressor. [47] Compared to the other BH3 proteins, little is known about BMF. However, BMF does exhibit certain features that are similar to BIM. [47] In response to certain stimuli, BIM, and sometimes BMF, may release from the cytoskeleton, translocate to the mitochondria, and neutralize the Bcl-2 anti-apoptotic molecules to trigger pro-apoptotic activation resulting in caspase-mediated cell killing [47].
BH3-only proteins bind selectively to the Bcl-2 pro-survival (anti-apoptotic) proteins BAD and BMF bind only to Bcl-2 Bcl-w and Bcl-xL, while NOXA binds only to Mcl-1 and A1. [37] Determining the selectivity will allow the development of BH3 mimetics that would target the anti-apoptotic proteins. [37] Proper control of cell death plays an important role in controlling the formation of memory T cells. [45] The deregulation of apoptosis in the mitochondrial or intracellular pathways severely impacts the abundance of activated memory T cells. [45] Studies of the BH3-only proteins reveal overlapping roles between the members in specific immune cell subsets [1].
The Bcl-2 family in Tumorigenesis
Somatic cells are all born through mitosis and will mostly die through apoptosis or programmed cell death. [52] This process is interrupted in cancer. Mutations that impair apoptosis can prompt cancer development and reduce the results of effective therapies. [54] Cancer may be induced when this delicate balance is disrupted by an increase in cell proliferation or a decrease in cell death. [52] Impaired apoptosis is central in cancer development and is a crucial barrier to effective treatment. [49] Bcl-2 family proteins play a central role in cell death regulation. Mutant Bcl-2 inhibits apoptosis that is usually triggered by many different internal and external stimuli. The imbalance of Bcl-2 proteins can induce cancer, the overgrowth of malignant cells [56].
Follicular lymphoma is a common human B-cell neoplasm. [56] Bcl-2 was the first member of the Bcl-2 family to be implicated in cancer through its involvement in the t14; 18 chromosome translocation. [50] Mice that mimicked the translocation confirmed that the deregulation of Bcl-2 expression is oncogenic, but the tumor incidence was low. [51] When Bcl-2 is expressed in cells in culture, it protects the cells from apoptosis after treatments from a diverse range of drugs and toxins, giving them antibody resistance. This infers that Bcl-2 not only has a hand in the development of malignant cells but also gives them resistance to therapy. Despite this, apoptosis resistance itself does not give rise to cancerous cells but combining the inhibition of apoptosis, like by Bcl-2, with a conventional growth stimulatory oncogene, like c-myc, malignant tissues can proliferate rapidly. [52] Mice that mimicked the translocation confirmed that the deregulation of Bcl-2 expression is oncogenic, but the tumor incidence was low. [51] Experiments in vitro demonstrated that starving Bcl-2 expressed cells inhibits cell death but does not promote cell proliferation. [3] However, Bcl-2 transgenic mice confirm that the inhibition of apoptosis induces cancer, as these mice became afflicted with cancer as they aged. [53] The long latency period reveals that additional oncogenic mutations are necessary to prompt the transition from low-grade polyclonal hyperplasia to monoclonal high-grade malignancy. [56] The Bcl-2 protein family regulates cell death and cell proliferation. Cell death and proliferation are dysregulated during oncogenic transformation. Therefore, we can conclude that the upregulation of Bcl-2 expression is in many cancers [55].
The development of tumors requires a combination of defects that prompts nascent neoplastic cells to become self-reliant for cell proliferation and resistant to signals that restrain cell growth. [56.] Research has demonstrated that cell death in normal adult tissues must continuously occur to balance cell proliferation to maintain homeostasis in healthy cells. [57] Defects in apoptosis, caused by the survival of unwanted cells or the inappropriate killing of vital cells, are considered the hallmarks of many disorders, including degenerative diseases, cancer, and autoimmune disorders [58].
In tumorigenesis, the overexpression of the prosurvival members of the Bcl-2 family has been found to be a common feature in many human cancers. [56] There is substantial evidence that the overexpression of Bcl-2 like proteins is critical in tumor development, maintenance, and resistance [61].
As mentioned before, the Bcl-2 protein is overexpressed in human follicular center B-cell lymphoma as a result of the chromosome translocation. [56,62] Bcl-2 has also been detected in significant levels in chronic lymphocytic leukemia (CLL) and mantle cell lymphoma. High levels of Bcl-2 expression were also detected, not only in hematological malignancies but also solid tumors in the brain, breasts, and lungs. [56,63] The high levels of Bcl-2 expression have been noted in CLL and other cancers possibly due to the loss of negative regulation of microRNA (miRNA) [64].
Compared to Bcl-2, Bcl-xL levels are lower, but it has been proposed that they promote the survival of follicular and germinal center B cells. [65] These cells are undergoing Ig class switch recombination and hypermutation of the variable regions of the rearranged Ig genes [65] and are targets for impaired DNA double-strand break that could cause chromosomal translocation. [66] Bcl-xL, like Bcl-2, cannot cause cancer on its own, as demonstrated in mice, but additional mutations, such as overexpression of Myc, could lead to the development of plasmacytoma. [66] Bcl-xL has been linked to the development and therapeutic resistance of Bcr/ Abl+ chronic myelogenous leukemia. [67] Signal pathways, activated by the Bcr/Abl oncogenic tyrosine kinase and the STAT5 transcription factor, activator of Bcl-x transcription, plays an important role in cellular transformation [67].
Mcl-1, identified as an early response gene, induces the differentiation of ML-1 human myeloblastic leukemia cells. [68] It has exhibited high levels in a range of hematological and solid malignancies, including cholangiocarcinomas, acute myeloid leukemia (AML) and multiple myeloma. [56] Forced expression of Mcl-1 inhibits Myc-induced apoptosis and synergizes with an Eμ-myc transgene in lymphomagenesis. [69] According to a study of 151 clinical isolates of non-Hodgkin's B lymphoma, high levels of Mcl-1 expression correlated with increasing severity in follicular lymphoma. [70] The BH3 mimetic ABT-737 that binds and inhibits Bcl-2, Bcl-xL, and Bcl-w, but not Mcl-1 was unable to stop the proliferation of B-cell lymphomas expressing high levels of Mcl-1. [71] Recent research has found that somatic cells that possess an increasing amount of Mcl-1 have been found in lung and breast cancers, concluding that Mcl-1 is critical for sustained survival and growth of these malignant cells. [72] These results signify that Mcl-1 is crucial to many malignant cell types, is essential for sustained survival, and seems to impose an obstacle in effective anticancer therapy [56].
Bfl-1 is also expressed in a range of hematopoietic cell types. Bfl-1 is expressed in B and T lymphocytes, neutrophils, mast cells, macrophages and dendritic cells. [73] In many of these cells, Bfl-1 expression is induced by the activation of antigen or cytokine receptors, but Bfl-1 levels also rapidly decline due to its rapid turnover. [74] A target for anticancer therapies, Bfl-1 overexpression is a signature point in certain B lymphoid malignancies. [75] A1, the murine homolog of human Bfl-1, has been implicated in the development of Bcr/Abl+ chronic myeloid leukemia (CML), like Bcl-xL [56].
To date, high levels of Bcl-w have not been linked to hematological malignancies; however; Bcl-2 overexpression has been reported to promote survival and influence the migratory and invasive potential of gastric cancer cells. [76] The study reports the Bcl-w expression, not Bcl-2 expression, correlated with the increasing levels of matrix metalloproteinase-2, an enzyme that affects the destruction of the basement membrane to facilitate metastasis. [56] Additionally, high levels of Bcl-w have been reported in advanced staged cancers rather than local tumors. [77] Based on the data collected, it is possible to conclude the role of Bcl-w relates to the spread of the malignant cells, rather than the formation.
Only a small portion of the cancers contain the chromosomal translocation that indicated the massive overexpression of antiapoptotic Bcl-2 family members. [56] Knowing what Bcl-2 proteins are critical in which type of tumor will provide clues for the development of cancer therapies.
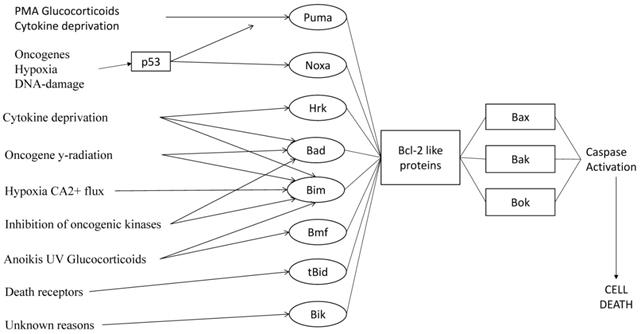
BH3-only proteins, a pro-apoptotic subgroup of the Bcl-2 family that is essential for apoptosis signaling, trigger apoptosis by binding via their BH3 region to a groove on the surface of the anti-apoptotic proteins. [11] (Figure 3) Studies have shown that different apoptotic stimuli require different BH3-only proteins for cell death. [60] PUMA is essential for p53-induced apoptosis prompted by DNA damage, anoxia or Myc overexpression. [80,81] Bim is critical for growth factor deprivation, deregulated calcium flux-induced killing, and ER stress in different cell types. [82] The combined loss of BIM and PUMA causes cell types to become more resistant to cytokine deprivation and other apoptotic stimuli, rather than the loss of either BH3-only protein alone. [81,84] BH3-only proteins act as an essential barrier against the development of malignant diseases, serving as molecular sentinels that kill abnormal cells that have potentially neoplastic lesions. [60] According to research, the loss of BIM [86] and PUMA [87], two key initiators of cytokine-induced apoptosis, inhibits the death of Myc-overexpressing B cells and accelerates Eμ-myc-induced lymphomagenesis. [88] The loss of BIM alleles has been found in 20% of human mantle B cell lymphoma. [89] About 40% of Burkitt lymphomas express extremely low levels of PUMA [87], potentially due to epigenetic silencing. [87] Also, the loss of a PUMA allele was found in a range of different human cancers [72], allowing us to conclude that the pro-apoptotic BH3-only members of the Bcl-2 family function as tumor suppressors.
Activation of BH3-only proteins through different stimuli. Different apoptotic stimuli activate different BH3-only members. Some stimuli activate multi BH3-only proteins. [38]
Ongoing Research
The high rate of apoptosis in basal cell carcinomas of the skin demonstrate that these are slow growing tumors, in spite of their high mitotic rate. [52] Current research is exploring the range of BH3 mimetics, drugs that mimic BH3 only proteins by binding to anti-apoptotic to block their function. [54] Clinical trials of venetoclax are underway for diverse cancers, and preclinical studies signal the potential of emerging BH3 mimetics that target other BCL-2 relatives, particularly MCL-1. Therefore, this new class of anticancer drugs may well find broad applications [54].
The ability to trigger cell apoptosis in tumors is responsible for the therapeutic effects of chemotherapeutic drugs novel cancer drugs, and radiation. The function of BIM, responsible for the killing of tumor cells by inhibitors of oncogenic kinases, such as the responses from CML to BCR-ABL inhibitor Gleevec, can be mimicked by ABT-737. [59] A closely related member of ABT-737, the ABT-263, which is designed to target the hydrophobic groove of Bcl-xL, can also bind to Bcl-2 and Bcl-w with very high affinity. [78] ABT-737, a BH3 mimetic, requires the activation of Bax/ Bak for cell killing, activating them indirectly by neutralizing the anti-apoptotic proteins, such as Bcl-2, Bcl-xL and Bcl-w. [71] Although the mimetic ABT-737 can bind and induce apoptosis, it is not effective at killing tumor cells that express high levels of Mcl-1, but it can effectively induce death in tumors when combined with other developing anticancer therapies [71,59].
A problem with the BH3-only mimetics is that they also affect non-transformed cells. The two mimetics, ABT-737 and ABT-263, could also cause thrombocytopenia because the survival of platelets also relies on Bcl-xL. [61] This problem can be circumvented using ABT-199, or venetoclax, a Bh3-mimetic that inhibits only the Bcl-2 gene. ABT-199 shows great promise in future treatments for CLL [83].
The anti-apoptotic proteins Mcl-1 and Bcl-2 have been shown to be essential in T cell development and in homeostasis. However, the precise mechanisms of how these proteins function in T cells and other cells are unclear. [43] In developing T cells, Mcl-1 has a unique role in supporting T cell development. It has been discovered that the developmental defects that arise from the deletion of Mcl-1 can be partially rescued by the deficiency Bak gene [79].
Further research has shown two opposing but not exclusive models for the system of the anti-apoptotic proteins, Bcl-2, Bcl-xL, and Mcl-1. The direct-activation model indicates the anti-apoptotic proteins inhibit the BH3-only activator proteins. By this model, certain Bh3-only molecules activate Bak and Bax directly and bind to the anti-apoptotic proteins to these Bh3-only activators which inhibit the apoptotic pathways.However, under the Bak/Bax sequestration model, the anti-apoptotic proteins bind to Bak and Bax directly to prevent their oligomerization or activation. [12] Based on this alternate model, Bh3- only proteins function to affect the ability of anti-apoptotic proteins to bind Bak/Bax and the distinction between the sensitizers and activators lie in the different abilities of the Bh3- only proteins that bind to anti-apoptotic proteins [85,34].
Conclusion
The Bcl-2 family of proteins is essential in the regulation of apoptosis. The delicate balance of pro-apoptotic and anti-apoptotic factors within the cell determines the fate of the cell. The overexpression of the prosurvival members of the Bcl-2 family inhibits cell death, leading to the prolonged survival of the cell. Combined with other oncogenic mutations, the cell could become malignant and attack neighboring cells. Tumors are formed through translocation of the Bcl-2 family and additional mutations, such as the myc gene.
The Bcl proteins are involved in the intrinsic apoptotic pathway, which is significant in the development of the immune system. The deletion of any gene of the Bcl-2 family can result in the abnormal development of lymphocytes. The balance of pro-apoptotic versus anti-apoptotic proteins, which operate at the organelle level, determines if the lymphocyte lives or dies. The Bcl-2 family of proteins could also induce the premature proliferation of the lymphocytes, which could often contain mutations. The premature proliferation of lymphocytes is the result of the unsettling balance of antiapoptotic and proapoptotic proteins.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ludwig LM, Nassin ML, Hadji A, LaBelle JL. Killing Two Cells with One Stone: Pharmacologic BCL-2 Family Targeting for Cancer Cell Death and Immune Modulation. Frontiers in Pediatrics. 2016;4:135. doi:10.3389/fped.2016.00135
2. Fukuhara S, Rowley JD. Chromosome 14 translocations in non-Burkitt lymphomas. International Journal of Cancer. 1978;22:14-21
3. Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335:440-42
4. Yip KW, Reed JC. Bcl-2 family proteins and cancer. Nature Oncogene. 2008;27:6398-406
5. Shamas-Din A, Brahmbhatt H, Leber B, Andrews DW. BH3-only proteins: Orchestrators of apoptosis. Biochimica et Biophysica Acta. 2011;1813:508-20
6. Tsujimoto Y. Role of Bcl-2 family proteins in apoptosis: apoptosomes or mitochondria? Genes Cells. 1998;3:697-707
7. Ahsen OV, Renken C, Perkins G, Kluck RM, Bossy-Wetzel E, Newmeyer DD. Preservation of Mitochondrial Structure and Function after Bid- or Bax-Mediated Cytochrome c Release. Journal of Cell Biology. 2000;150:1027-36
8. Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, Green DR. Direct Activation of Bax by p53 Mediates Mitochondrial Membrane Permeabilization and Apoptosis. Science. 2004;303(5660):1010-14
9. Kang MH, Reynolds CP. Bcl-2 Inhibitors: Targeting Mitochondrial Apoptotic Pathways in Cancer Therapy. Clinical Cancer Research. 2009;15:1126-32
10. Thomas S, Quinn BA, Das SK. et al. Targeting the Bcl-2 Family for Cancer Therapy. Expert Opinion On Therapeutic Targets. 2013;17:61-75
11. Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nature Reviews Molecular Cell Biology. 2008;9:47-59
12. Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Kormeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183-9
13. Nakan K, Vousden KH. PUMA, a novel proapoptotic gene, is Induced by p53. Molecular Cell. 2001;7:683-94
14. Certo M, Del Gaizo Moore V, Nishino M, Wei G, Kormeyer S, Armstrong SA, Letai A. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351-65
15. Li P. et al. Cytochrome c and dATP-dependent formation of Apaf-1/Caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479-89
16. Hakem R. et al. Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell. 1998;94:339-52
17. Lindsten T, Ross AJ, King A. et al. The combined functions of proapoptotic bcl-2 family members Bak and Bax are essential for normal development of multiple tissues. Molecular cell. 2000;6:1389-99
18. Biswas S, Shi Q, Matise L, Cleveland S, Dave U, Zinkel S. A role for proapoptotic Bax and Bak in T-cell differentiation and transformation. Blood. 2010;116:5237-46
19. Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1998;335:440-2
20. Strasser A, Whittingham S, Vaux DL. et al. Enforced BCL2 expression in B-lymphoid cells prolongs antibody responses and elicits autoimmune disease. Proceedings of the National Academy of Sciences. 1991;88:8661-5
21. Veis DJ, Sorenson CM, Shutter JR, Korsmeyer SJ. Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell. 1993;75:229-40
22. Rinkenberger JL, Horning S, Klocke B, Roth K, Korsmeyer SJ. Mcl-1 deficiency results in peri-implantation embryonic lethality. Genes and Development. 2000;14:23-7
23. Kozopas KM, Yang T, Buchan HL, Zhou P, Craig RW. MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to Bcl-2. Proceedings of the National Academy of Sciences. 1993;90:3516-20
24. Zhou P, Qian L, Kozopas KM, Craig RW. Mcl-1, a Bcl-2 family member, delays the death of hematopoietic cells under a variety of apoptosis-inducing conditions. Blood. 1997;89:630-43
25. Opferman JT, Iwasaki H, Ong CC, Suh H, Mizuno S, Akashi K, Korsmeyer SJ. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science. 2005;307:1101-4
26. Opferman JT, Letai A, Beard C, Sorcinelli MD, Ong CC, Korsmeyer SJ. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature. 2003;426:671-6
27. Pierson W, Cauwe B, Policheni A. et al. Anti-apoptotic Mcl-1 is critical for the survival and niche-filling capacity of Foxp3+ regulatory T cells. Nature immunology. 2013;14:959-65
28. Vikstrom I, Carotta S, Lüthje K. et al. Mcl-1 is Essential for Germinal Center Formation and B cell Memory. Science. 2010;330:1095-9
29. Peperzak V, Vikström I, Walker J. et al. Mcl-1 is essential for the survival of plasma cells. Nature immunology. 2013;14:290-7
30. Ma A, Pena JC, Chang B. et al. Bclx regulates the survival of double-positive thymocytes. Proceedings of the National Academy of Sciences. 1995;92:4763-7
31. Herold MJ, Stuchbery R, Mérino D. et al. Impact of conditional deletion of the pro-apoptotic BCL-2 family member BIM in mice. Cell Death & Disease. 2014;5:e1446. doi:10.1038/cddis.2014.409
32. Tsukamoto H, Huston GE, Dibble J, Duso DK, Swain SL. Bim dictates naïve CD4 T cell lifespan and the development of age-associated functional defects. Journal of immunology. 2010;185:4535-44
33. Ren D, Tu H-C, Kim H. et al. BID, BIM, and PUMA Are Essential for Activation of the BAX- and BAK-Dependent Cell Death Program. Science. 2010;330:1390-3
34. Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L, Czabotar PE, Ierino H. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science. 2007;315:856-9
35. Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA induces the rapid apoptosis of colorectal cancer cells. Molecular Cell. 2001;7:673-82
36. Opferman JT, Korsmeyer SJ. Apoptosis in the development and maintenance of the immune system. Nature Immunology. 2003;4:410-5
37. Willis SN, Adams JM. Life in the balance: how BH3-only proteins induce apoptosis. Current Opinion in Cell Biology. 2005;17:617-25
38. Korsmeyer SJ, Wei MC, Saito M, Weiler S, Oh KJ, Schlesinger PH. Pro-apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ. 2000;7:1166-73
39. Clybouw C, Fischer S, Auffredou MT. et al. Regulation of memory B-cell survival by the BH3-only protein Puma. Blood. 2011;118:4120-8
40. Garrison SP, Thornton JA, Häcker H. et al. The p53-Target Gene Puma Drives Neutrophil-Mediated Protection against Lethal Bacterial Sepsis. PLoS Pathogens. 2010;6:e1001240. doi:10.1371/journal.ppat.1001240
41. Strasser A. The role of BH3-only proteins in the immune system. Nature Reviews Immunology. 2005;5:189-200
42. Pradhan S, Genebriera J, Denning WL, Felix K, Elmets CA, Timares L. CD4 T cell-induced, Bid-dependent apoptosis of cutaneous dendritic cells regulates T cell expansion and immune responses. Journal of immunology (Baltimore, Md : 1950). 2006;177(9):5956-5967
43. Dunkle A, Dzhagalov I, He Y-W. Mcl-1 promotes survival of thymocytes by inhibition of Bak in a pathway separate from Bcl-2. Cell death and differentiation. 2010;17:994-1002
44. Wensveen FM, Klarenbeek PL, van Gisbergen KP, Pascutti MF, Derks IA, van Schaik BD, Ten Brinke A, de Vries N, Cekinovic D, Jonjic S, van Lier RA, Eldering E. Pro-apoptotic protein Noxa regulates memory T cell population size and protects against lethal immunopathology. J Immunol. 2013;190:1180-91
45. Sabbagh L, Srokowski CC, Pulle G. et al. A critical role for TNF receptor-associated factor 1 and Bim down-regulation in CD8 memory T cell survival. Proceedings of the National Academy of Sciences. 2006;103:18703-8
46. Ranger AM, Zha J, Harada H. et al. Bad-deficient mice develop diffuse large B cell lymphoma. Proceedings of the National Academy of Sciences. 2003;100:9324-9
47. Labi V, Erlacher M, Kiessling S. et al. Loss of the BH3-only protein Bmf impairs B cell homeostasis and accelerates γ irradiation-induced thymic lymphoma development. The Journal of Experimental Medicine. 2008;205:641-55
48. Delbridge ARD, Strasser A. The BCL-2 protein family, BH3-mimetics and cancer therapy. Cell Death and Differentiation. 2015;22:1071-80
49. Farbman AI. Electron microscope study of palate fusion in mouse embryos☆. (n.d.). Developmental Biology. 1968;18:93-116
50. Adams JM, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene. 2007;26:1324-37
51. McDonnell TJ, Korsmeyer SJ. Progression from lymphoid hyperplasia to high-grade malignant lymphoma in mice transgenic for the t(14; 18). Nature. 1991;349:254-6
52. Gerl R, Vaux DL. Apoptosis in the development and treatment of cancer. Carcinogenesis. 2005;26:263-70
53. Strasser A, Harris AW, Bath ML, Cory S. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature. 1990;348:331-3
54. Cory S. et al. Targeting BCL-2-like Proteins to Kill Cancer Cells. Trends in Cancer. 2016;2:443-60
55. Akl H, Vervloessem T, Kiviluoto S, Bittremieux M, Parys JB, De Smedt H, Bultynck G. Biochimica et Biophysica Acta. 2014. 1843:2240-52
56. Kelly PN, Strasser A. The role of Bcl-2 and its pro-survival relatives in tumourigenesis and cancer therapy. Cell Death and Differentiation. 2011;18:1414-24
57. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. British Journal of Cancer. 1972;26:239-57
58. Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death in disease: mechanisms and emerging therapeutic concepts. The New England Journal of Medicine. 2009;361:1570-83
59. Kuroda J, Puthalakath H, Cragg MS. et al. Bim and Bad mediate imatinib-induced killing of Bcr/Abl+ leukemic cells, and resistance due to their loss is overcome by a BH3 mimetic. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:14907-12
60. Huang DC, Strasser A. BH3-only proteins - essential initiators of apoptotic cell death. Cell. 2003;103:839-42
61. Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J. et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nature Medicine. 2013;9:202-8
62. Cleary ML, Sklar J. Nucleotide sequence of a t(14; 18) chromosomal breakpoint in follicular lymphoma and demonstration of a breakpoint-cluster region near a transcriptionally active locus on chromosome 18. Proceedings of the National Academy of Sciences. 1985;82:7439-43
63. Castle VP, Heidelberger KP, Bromberg J, Ou X, Dole M, Nuñez G. Expression of the apoptosis-suppressing protein bcl-2, in neuroblastoma is associated with unfavorable histology and N-myc amplification. The American Journal of Pathology. 1993;143:1543-50
64. Cimmino A, Calin GA, Fabbri M. et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proceedings of the National Academy of Sciences. 2005;102:13944-9
65. Didier AM. et al. bcl-x exhibits regulated expression during B cell development and activation and modulates lymphocyte survival in transgenic mice. The Journal of Experimental Medicine. 1996;183:381-91
66. Cheung WC, Kim JS, Linden M. et al. Novel targeted deregulation of c-Myc cooperates with Bcl-XL to cause plasma cell neoplasms in mice. Journal of Clinical Investigation. 2004;113:1763-73
67. Horita M, Andreu EJ, Benito A. et al. Blockade of the Bcr-Abl kinase activity induces apoptosis of chronic myelogenous leukemia cells by suppressing signal transducer and activator of transcription 5-dependent expression of Bcl-XL. The Journal of Experimental Medicine. 2000;191:977-84
68. Kozopas KM, Yang T, Buchan HL, Zhou P, Craig RW. MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proceedings of the National Academy of Sciences. 1993;90:3516-20
69. Campbell KJ, Bath ML, Turner ML. et al. Elevated Mcl-1 perturbs lymphopoiesis, promotes transformation of hematopoietic stem/progenitor cells, and enhances drug resistance. Blood. 2010;116:3197-207
70. Cho-Vega JH, Rassidakis GZ, Admirand JH, Oyarzo M, Ramalingam P, Paraguya A, Medeiros LJ. MCL-1 expression in B-cell non-Hodgkin's lymphomas. Human Pathology. 2004;35:1095-100
71. Van Delft MF, Wei AH, Mason KD. et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer cell. 2006;10:389-99
72. Beroukhim R, Mermel CH, Porter D. et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899-905
73. Lin EY, Orlofsky A, Berger MS, Prystowsky MB. Characterization of A1, a novel hemopoietic-specific early-response gene with sequence similarity to bcl-2. The Journal of Immunology. 1993;151:1979-88
74. Xiang Z, Ahmed AA, Möller C, Nakayama K, Hatakeyama S, Nilsson G. Essential Role of the Prosurvival bcl-2 Homologue A1 in Mast Cell Survival After Allergic Activation. The Journal of Experimental Medicine. 2001;194:1561-70
75. Feuerhake F, Kutok JL, Monti S, Chen W, LaCasce AS, Cattoretti G, Shipp MA. NFκB activity, function, and target-gene signatures in primary mediastinal large B-cell lymphoma and diffuse large B-cell lymphoma subtypes. Blood. 2005;106:1392-9
76. Bae IH, Park M, Yoon SH, Kang SW, Lee S, Choi K, Um H. Bcl-w promotes gastric cancer cell invasion by inducing matrix metalloproteinase-2 expression via phosphoinositide 3-kinase, Akt, and Sp1. Cancer Research. 2006;66:4991-5
77. Wilson JW, Nostro MC, Balzi M. et al. Bcl-w expression in colorectal adenocarcinoma. British Journal of Cancer. 2000;82:178-85
78. Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC. et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677-81
79. Zhan Y, Carrington EM, Zhang Y, Heinzel S, Lew AM. Life and death of activated t cells: how are they different from naïve t cells? Frontiers in Immunology. 2017;8:1809. doi:10.3389/fimmu.2017.01809
80. Villunger A, Michalak EM, Coultas L, Müllauer F, Böck G. et al. p53- and drug-induced apoptotic responses mediated by BH3-only proteins Puma and Noxa. Science. 2003;302:1036-8
81. Erlacher M, Labi V, Manzl C. et al. Puma cooperates with Bim, the rate-limiting BH3-only protein in cell death during lymphocyte development, in apoptosis induction. The Journal of Experimental Medicine. 2006;203:2939-51
82. Puthalakath H, O'Reilly LA. et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007;129:1337-49
83. Adams J, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene. 2007;26:1324-37
84. Erlacher M, Michalak EM, Kelly PN. et al. BH3-only proteins Puma and Bim are rate-limiting for γ-radiation- and glucocorticoid-induced apoptosis of lymphoid cells in vivo. Blood. 2005;106:4131-8
85. Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day C, Adams J, Huang DC. Differential targeting of prosurvival bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol. Cell. 2005;17:393-403
86. Egle A, Harris AW, Bouillet P, Cory S. Bim is a suppressor of Myc-induced mouse B cell leukemia. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(16):6164-6169
87. Garrison SP, Jeffers JR, Yang C. et al. Selection against PUMA Gene Expression in Myc-Driven B-Cell Lymphomagenesis. Molecular and Cellular Biology. 2008;28(17):5391-5402 doi:10.1128/MCB.00907-07
88. Bouillet P, Metcalf D, Huang DC. et al. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 1999;286:1735-8
89. Tagawa H, Karnan S, Suzuki R, Matsuo K. et al. Genome-wide array-based CGH for mantle cell lymphoma: identification of homozygous deletions of the proapoptotic gene BIM. Oncogene. 2005;24:1348-58
90. Roset R, Gil-Gomez G, Ortet L. Role of the Bcl-2 family members on apoptosis: what we have learned from knock-out mice. Frontiers in Bioscience. 2007;12:4722-30
Author contact
![]() Corresponding author: Yi Lu, Ph.D., Department of Pathology and Laboratory Medicine, University of Tennessee Health Science Center, Cancer Research Building, 19 South Manassas Street, Memphis, TN 38163 (USA). Tel.: (901) 448-5436; Fax: (901) 448-5496; E-mail: yluedu
Corresponding author: Yi Lu, Ph.D., Department of Pathology and Laboratory Medicine, University of Tennessee Health Science Center, Cancer Research Building, 19 South Manassas Street, Memphis, TN 38163 (USA). Tel.: (901) 448-5436; Fax: (901) 448-5496; E-mail: yluedu